




El síndrome de activación de macrófago es una complicación inusual pero potencialmente fatal de pacientes con enfermedades reumáticas autoinmunes. Esta es una entidad clínico-patológica caracterizada por la activación de histiocitos con hemofagocitosis prominente en la médula ósea y otros sistemas reticuloendoteliales. En pacientes con lupus, puede simular una exacerbación de la enfermedad o infección. Presentamos el caso de una paciente de 7 años de edad en la que el diagnóstico de lupus eritematoso sistémico y síndrome de activación de macrófago fue simultáneo con respuesta al uso de ciclofosfamida.
The macrophage activation syndrome is a rare but potentially fatal complication of patients with autoimmune rheumatic diseases. This is a clinicopathological entity characterized by activation of histiocytes with prominent hemophagocytosis in the bone marrow and other reticuloendothelial systems. In patients with lupus it may mimic an exacerbation of the disease or infection. We report the case of a 7-year-old girl in whom the diagnosis of lupus erythematosus and macrophage activation syndrome was simultaneously made with response to the use of cyclophosphamide.
Las enfermedades reumáticas autoinmunes en pediatría son poco frecuentes, con una presentación clínica variada. Los niños representan del 15 al 20% del total de los pacientes con lupus eritematoso sistémico (LES), con una incidencia de 0,3 a 0,9 por 100.000 niños y una prevalencia de 3,3 a 8,8 por 100.000 niños1. A diferencia de los adultos, los niños tienen una enfermedad más severa al momento del diagnóstico2. Una de las complicaciones observadas en pacientes con LES es el síndrome de activación de macrófago (MAS). El MAS es una condición severa y potencialmente fatal asociada a una excesiva activación y expansión de macrófagos y células T con hipercitocinemia masiva, dando lugar a una respuesta inflamatoria descontrolada, con valores elevados de factor estimulante de colonias de macrófagos (M-CSF), TNF-α, IL-1β, IL-18, IL-6 e interferón gamma, con disminución en la expresión de perforina en células NK y linfocitos CD8. En el perfil de citocinas de pacientes con MAS asociado a LES, predominan el TNF-α y el M-CSF3–6. Sin embargo, el MAS no es una complicación frecuente pero presenta un curso rápido, por lo que su diagnóstico y tratamiento tempranos son necesarios. Su incidencia ha ido aumentando en pacientes con enfermedades reumáticas especialmente la asociada a LES.
La presentación clínica del MAS es generalmente aguda, requiriendo de la admisión a la Unidad de Cuidados Intensivos, con fiebre alta que no remite, pancitopenia, hepatomegalia, linfadenopatías generalizadas y elevación de enzimas hepáticas, pudiéndose observar en algunos casos alteraciones en las pruebas de coagulación, elevación de ferritina sobre 500 a 10.000ng/ml y disfunción de SNC en un tercio de los casos7. La característica patognomónica se encuentra en el aspirado de médula ósea (AMO), que muestra numerosos histiocitos morfológicamente benignos con actividad hemofagocítica. Su diagnóstico es un reto para el médico, ya que puede imitar recaídas o complicaciones infecciosas8.
Ante la falta de estudios controlados sobre tratamiento del MAS, este se basa en experiencias anecdóticas. Usualmente, se utilizan dosis altas de esteroides. La ciclosporina A (CsA) se utilizó a mediados de los noventa para las formas congénitas del síndrome hemofagocítico (SH) y, posteriormente, mostró efectividad en pacientes con MAS refractario a esteroides, por lo que se considera como terapia de primera línea9. Otros tratamientos, como la inmunoglobulina por vía intravenosa (IGIV), ciclofosfamida (CFM), anti-TNF, anakinra, plasmaféresis y etopósido, han mostrado resultados contradictorios.
Hasta la fecha, se han reportado 94 casos de MAS asociado a LES10; de estos, 60 son en pacientes pediátricos, con presentación simultánea de LES y MAS en 9 niños4,11–17.
La mortalidad reportada del MAS asociado a LES juvenil es del 11% y la disfunción orgánica es más común, comparado con otras enfermedades reumáticas3.
Presentamos el caso de una paciente pediátrica que presentó MAS asociado a las manifestaciones iniciales de LES, con falta de respuesta a esteroides, IGIV y CsA, que finalmente respondió con el uso de CFM.
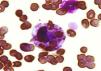
Caso clínicoNiña de 7 años de edad, evolución de 4 meses con astenia, adinamia e hiporexia, edema facial bipalpebral y dolor en el hipocondrio derecho, con progresión del edema hasta llegar a anasarca, disnea de grandes esfuerzos e hipertensión, por lo acude al hospital; se realizaron exámenes de laboratorio; se diagnosticó síndrome nefrótico/nefrítico, por lo que se envió a nuestro hospital. A su ingreso, presentó palidez generalizada, fiebre de 40°C, petequias, artritis de manos y codos, derrame pleural bilateral, soplo holosistólico 3/6, ascitis y hepatoesplenomegalia. La biometría hemática mostró leucocitos 1980/μL (VN 4.000-13.500/μL), neutrófilos 820/μL, linfocitos 910/μL, hemoglobina (Hb) 6,3g/dL (VN 11,5-14,5g/dL), plaquetas 44.000/μL (VN 150.000-400.000/μL), tiempo de tromboplastina parcial activado prolongado sin corrección tras la adición de plasma normal 1:2 (testigo 30 s), fibrinógeno 368mg/dL (VN 200-400mg/dL), anticuerpos antinucleares por CLIA 5 (VN 0,5-1,5), por IFI 1: 160 patrón homogéneo y anticuerpos anti-ADN 82 (VN 0-20), C3 38mg/dL (VN 90-177mg/dL), C4 3,3mg/dL (VN 15-45mg/dL), anticoagulante lúpico positivo con veneno de víbora de Russell y anticardiolipina IgG más de 280 μ/ml e IgM 162 μ/ml (positivo alto), test de serología luética falso positivo, anticuerpos treponémicos fluorescentes negativo, creatinina sérica de 1,4mg/dL y depuración de creatinina 34ml/min/1,73m2 (VN 70-150ml/min/1,73m2) y proteína C reactiva 50,4mg/L (VN 0-5mg/L). Con estos datos, se hizo el diagnóstico de LES con afectación renal, cutánea, articular y hematológica, con síndrome de antifosfolipídico secundario. Se inició tratamiento con pulsos de metilprednisolona (30mg/kg/día) por 5 días, con pobre respuesta; persistió con afección de las 3 líneas celulares en la biometría hemática, con elevación de triglicéridos a 287mg/dL (VN 120-200mg/dL). Ante la falta de respuesta a esteroide, se inició IGIV a dosis de 400mg/kg/día por 5 días y filgrastim; las pruebas de laboratorio mostraron leucocitos de 1.890/μL, neutrófilos de 0/μL, linfocitos 1.760/μL, plaquetas 57.000/μL, Hb 7,2g/dL con prueba de Coombs directo negativa. Se descartó proceso infeccioso con el panel viral para citomegalovirus, herpes 1 y 2, rubéola, virus de Epstein-Barr, toxoplasma, virus de la inmunodeficiencia humana, virus de las hepatitis B y C, y los cultivos bacterianos, que resultaron negativos. El AMO mostró disminución de la celularidad y gran cantidad de histiocitos, en conglomerado en todo el frotis, con fagocitosis de eritrocitos y plaquetas, haciéndose el diagnóstico de SH (fig. 1). La ferritina sérica fue de 1.058ng/ml. Se inició CsA a dosis de 1mg/kg/día (insuficiencia renal), por 4 días, sin respuesta, con leucocitos de 660/μL, neutrófilos 0/μL, linfocitos 580/μL y plaquetas 52.000/μL, por lo que se indicó CFM a 1g/m2 de superficie corporal, con dosis mensual. A los 15 días de la primera dosis de CFM, se observó franca recuperación hematológica, con leucocitos de 14.970/μL, neutrófilos 12.800/μL (en ausencia de infección), linfocitos 490/μL, Hb 11,6g/dL y plaquetas 90.000/μL. Durante su evolución, no ameritó uso de aminas vasopresoras o ventilación mecánica. Se egresó a su domicilio con prednisona a 1mg/kg/día y pulsos de CFM mensual. Tres meses después se realizó biopsia renal que mostró nefritis lúpica clase iv de la Organización Mundial de la Salud. Después de 14 pulsos mensuales de CFM, presentó síndrome de Fisher Evans (anemia hemolítica autoinmune y trombocitopenia inmune primaria) y neuropatía periférica, tratándose con esteroide en pulsos, IGIV, CFM y rituximab. Cumplió en total 18 pulsos mensuales de CFM y continuó tratamiento con micofenolato de mofetilo y esteroide a dosis bajas. La paciente, después de 3 años, lleva una vida normal y acude a la escuela.
Discusión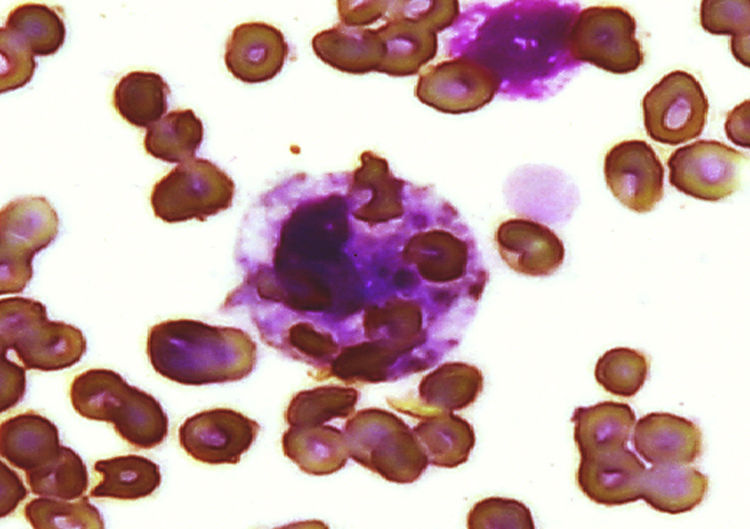
El SH es una complicación inusual pero potencialmente fatal en pacientes con enfermedades reumáticas autoinmunes. Esta es una entidad clínico-patológica que se caracteriza por la activación de histiocitos con hemofagocitosis prominente en la médula ósea y otros sistemas reticuloendoteliales18. Se presenta en formas primarias y secundarias. La forma primaria engloba un grupo de enfermedades genéticas que resultan en una función alterada de las células inmunitarias. La forma secundaria o adquirida se observa asociada a malignidad, infecciones o enfermedades autoinmunes8. En pacientes con enfermedades autoinmunes se denomina MAS y puede presentarse como manifestación inicial en el momento del diagnóstico o formar parte de una exacerbación o un proceso infeccioso4. Las enfermedades autoinmunes asociadas a MAS son la artritis reumatoide, el síndrome de Sjögren, la dermatomiositis, la enfermedad de Kawasaki, la esclerosis sistémica, la enfermedad mixta del tejido conjuntivo (EMTC) y el LES18. A pesar de no existir datos específicos sobre la prevalencia de MAS en el LES, se estima que va del 0,9 al 4,6%7.
El diagnóstico de MAS es difícil, ya que puede simular una complicación infecciosa o una exacerbación de la enfermedad de base18. Presentamos el caso de una paciente con diagnóstico simultáneo de LES y MAS. Cuando se presentan de manera paralela, la dificultad para realizar el diagnóstico aumenta, ya que las manifestaciones clínicas de ambas entidades se solapan.
La paciente cumplió con los criterios del American College of Rheumatology para LES y los criterios para MAS de la Histiocyte Society8, de Kamakura et al.19, y los criterios que reportaron recientemente Parodi et al.7. Los criterios de la Histiocyte Society tienen alta especificidad pero la sensibilidad no es satisfactoria, ya que aproximadamente el 33% de los pacientes con MAS no cumplen con esos criterios7. En 2004, Kamakura et al. indicaron criterios diagnósticos para el SH asociado a enfermedades autoinmunes, ya que las manifestaciones clínicas son distintas de las de otros síndromes hemofagocíticos reactivos19. En 2009, Parodi et al. elaboraron unas guías preliminares para el diagnóstico de MAS en pacientes con LES juvenil utilizando 5 criterios clínicos (fiebre, hepatomegalia, esplenomegalia, manifestaciones hemorrágicas y disfunción del SNC) y 6 de laboratorio (citopenias que afectan a 2 o más líneas celulares, aumento de la aspartato transaminasa, DHL, triglicéridos y ferritina, disminución de fibrinógeno) y el criterio histopatológico (evidencia de hemofagocitosis). Con uno o más criterios clínicos y 2 o más de laboratorio, se puede establecer el diagnóstico, el AMO se reserva para los casos dudosos. De los criterios de laboratorio, el que mostró mayor sensibilidad (96%) y especificidad (100%) fue la hiperferritinemia7.
En la revisión sistemática realizada por Atteritano et al. de SH en enfermedades reumáticas, encontraron a 94 pacientes con LES y SH10. Una revisión más detallada de los casos asociados a LES mostró que de los 94 casos, 60 correspondían a pacientes pediátricos y en 29 la presentación de LES y SH fue simultánea, 9 de estos en pacientes pediátricos. Nuestro caso sería el décimo caso con diagnóstico simultáneo reportado en población pediátrica y el de menor edad.
La revisión de los casos pediátricos en los que la presentación de LES y MAS fue simultánea, incluyendo el nuestro, se muestran en la tabla 14,11-17. Encontramos que la fiebre, las citopenias de al menos 2 líneas celulares y la hemofagocitosis se presentaron en el 100% de los casos, la hiperferritinemia en 70% de los casos (en 30% no se encontró disponible el dato al tratarse de series de casos), la esplenomegalia y la hipofibrinogenemia en el 50%. La hemofagocitosis se demostró en médula ósea en el 100%, ganglio linfático en el 30%, hígado en el 20% y bazo en el 10%.
Características de los pacientes reportados en la literatura con diagnóstico simultáneo de LES y MAS
| Número de referencia | Edad (años) y sexo | Fiebre | Citopenia de 2 o más líneas | Esplenomegalia | Hipertrigliceridemia | Hipofibrinogenemia | Hiperferritinemia | Hemofagocitosis | Anticuerpos | Afección de LES | Tratamiento | Resultado | SLEDAI |
| 4 | F 15 | X | Pancitopenia | – | ND | X | X | MOBazoHígadoGanglios | ANA 1:1280Anti-ADNAnti-RNP | HematológicaRenal | MPDIGIVEtopósidoCSA | Muerte | ND |
| 11 | F 11 | X | X | X | – | X | ND | GangliosMO | Anti-ADNANA positivo | CutáneaPulmonarRenal | ND | Vivo | ND |
| 12 | F 14 | X | Pancitopenia | ND | ND | ND | X | MO | ANA 1:1.280 | RenalHematológica | MPD IGIVCSA | Viva | ND |
| 13 | F 11 | X | Pancitopenia | – | ND | X | X | MO | ANA 4 +Anti-ADN 4+ | CutáneoRenalPancreatitis | MPDCSACFM | Viva | ND |
| 14 | F 11 | X | LeucopeniaTrombocitopenia | ND | – | X | ND | MOGanglio | ANA+1:1.000 | PulmonarCutáneoMiocarditisArticularRenalHematológico | MPD | Vivo | 21 |
| 14 | M 17 | X | Pancitopenia | X | – | – | X | MOHígado | ANA+1:1.280 | PulmonarRenalArticularCutáneoHematológico | MPDIGIVEtopósidoCsACFM+RTX | Vivo | 29 |
| 15 | F 17 | X | Bicitopenia | X | ND | ND | ND | MO | ND | ND | ND | ND | ND |
| 16 | F 10 | X | Pancitopenia | X | ND | X | X | MO | ANA 1:2.560Anti-ADNACL IgG + | RenalPancreatitisHematológico | EsteroidePlasmaféresisCFM | Vivo | ND |
| 17 | F 11 | X | LeucopeniaTrombocitopenia | ND | ND | X | MO | ANA 1:1280Anti-ADN 160 positivo | RenalCutáneoHematológico | MPDIGIV | Viva | ND | |
| Nuestro caso | F 7 | X | Pancitopenia | X | X | – | X | MO | ANA 1:160CLIA 5 positivoAnti-ADN +LA +ACL IgG e IgM + | RenalCutáneoArticularHematológico | MPDIGIVCsACFM | Viva | 23 |
ACL: anticardiolipinas; Anti-ADN: anticuerpos anti-ADN; ANA: anticuerpos antinucleares; CFM: ciclofosfamida; CsA: ciclosporina A; IgG: inmunoglobulina G; IgM:inmunoglobulina M; IGIV: inmunoglobulina por vía intravenosa; LA: anticoagulante lúpico; LES: lupus eritematoso sistémico; MO: médula ósea; MPD: metilprednisolona; ND: no disponible; RTX rituximab.
Los síntomas principales del MAS son la fiebre alta prolongada, hepatoesplenomegalia y citopenias. La presencia de citopenias es frecuente en el LES, pero la pancitopenia solo ocurre en menos del 10% de los casos y si se presenta, por lo general, es leve. La leucopenia se detecta de manera similar en pacientes con actividad de LES y MAS; sin embargo, cuando se encuentra leucopenia severa con cifras menores de 2.000 células, se debe considerar la presencia de MAS. La trombocitopenia es el mejor indicador de MAS comparado con leucopenia y anemia7,19. En los casos reportados de LES juvenil y MAS simultáneos, la pancitopenia se presentó en el 60%, la leucopenia y la trombocitopenia en el 80%. El caso que presentamos evolucionó a una leucopenia menor de 2.000 células; lo que llamó la atención fue la neutropenia, que llegó a ser absoluta, siendo datos atípicos a la presentación habitual de LES juvenil, lo que puede ser un indicador de alteración primaria de la médula ósea y orientar al diagnóstico de MAS.
La revisión realizada por Parodi et al.7 en 38 pacientes con LES juvenil y MAS comparados con un grupo de 29 pacientes con LES activo y un grupo de 387 pacientes de una cohorte de daño de LES, mostró que los pacientes que desarrollaron MAS tuvieron una frecuencia elevada de nefritis, artritis, serositis y afección hematológica en el momento del diagnóstico de MAS. En los 10 pacientes pediátricos con diagnóstico simultáneo de LES y MAS, se encontró afección renal en el 90%, hematológica en el 70%, cutánea en el 60%, pulmonar y articular en el 30% y cardiaca en el 10%, y llama la atención la afección pancreática en el 20%, ya que esta es una afección poco frecuente de LES pediátrico y en la literatura solo hay reportes de casos y pequeñas series. En el estudio realizado por Wang et al. en pacientes adultos y pediátricos con LES y pancreatitis, se encontró que el SLEDAI promedio de los pacientes pediátricos en el momento de la pancreatitis fue de 21, por lo que una actividad elevada de la enfermedad pudiera relacionarse con la aparición de MAS20. Apoyando lo anterior en la serie reportada por Lambotta de 15 casos de MAS asociado a LES, el promedio de SLEDAI fue 2214. Kim et al. realizaron un estudio de casos y controles de 15 pacientes con LES asociado a MAS, comparándolos con pacientes con LES sin MAS, encontrando en el primer grupo un SLEDAI más alto (14 vs. 8) y citopenias más profundas, siendo estadísticamente significativo21. De los 10 casos con diagnóstico simultaneo de LES y MAS, en los 2 pacientes en que se reportó SLEDAI este fue mayor de 20 y en nuestra paciente fue de 23. Tomando en conjunto estas observaciones, indicamos que un valor elevado de SLEDAI con afección hematológica y renal puede ser un factor de riesgo para el desarrollo de MAS.
La estrategia terapéutica no está bien establecida en el MAS que complica el LES. Como las infecciones son un desencadenante común, su exclusión es importante para establecer un adecuado tratamiento. En caso de infección, se debe disminuir la dosis de inmunosupresores. Como terapias de soporte, se utilizan IGIV y FECG en caso de neutropenia18.
La terapia inmunosupresora está indicada cuando el factor desencadenante es la actividad de la enfermedad. Bennet et al.y Parodi et al. en sus estudios encontraron que los tratamientos utilizados son esteroides a dosis altas, IGIV, CsA, CFM, micofenolato de mofetilo, azatioprina, rituximab, plasmaféresis o anti-TNF2,3,21. En la revisión de los casos pediátricos con diagnóstico simultáneo de LES y MAS, se utilizaron esteroides en el 100% de los casos, IGIV, CsA en el 50%, CFM en el 40% y rituximab y plasmaféresis en el 10%. La respuesta favorable con el uso de esteroides solos fue del 10%, con CsA del 40%, con CFM 100% (un caso asociado a rituximab) e IGIV 20%. Nuestra paciente fue tratada con esteroides a dosis altas, IGIV y CsA, sin obtener respuesta, por lo que finalmente se trató con CFM, obteniendo una respuesta exitosa. Es importante recalcar que la pancitopenia no empeoró con el uso de CFM. Aunque este tratamiento ha mostrado resultados contradictorios9; basándonos en la efectividad mostrada en estos casos se puede suponer que la CFM es una opción terapéutica que debe tomarse en cuenta en el tratamiento del MAS asociado al inicio del LES.
Para casos refractarios, se han utilizado también la terapia anti-TNF. Kikuchi et al.22 y Takahashi et al.23 reportaron 2 casos refractarios a terapia con esteroide, IGIV, CsA y en un caso metotrexato que respondieron con etanercept. Ideguchi et al.24 y Henzan et al.25 reportaron 2 casos refractarios a esteroides, ciclosporina, plasmaféresis y etopósido, que finalmente respondieron con infliximab; a uno de ellos se le trató con CFM pero una sola dosis a 500mg, por lo que quizá no se obtuvo la respuesta deseada. Nuestra paciente respondió a dosis de 1 gr/m2 de superficie corporal por vía intravenosa sin efectos inmediatos indeseables (pancitopenia profunda).
Otra opción terapéutica es anakinra, que mostró utilidad en pacientes con MAS asociado a vasculitis y artritis idiopática juvenil sistémica; en estas enfermedades, en el perfil de citocinas predominan la IL-1, la IL-6 y la IL-1826-28. Debido que el tratamiento del MAS se enfoca también al tratamiento de la enfermedad subyacente y en pacientes con MAS asociado a LES el perfil de citocinas es distinto, el uso de CFM en nuestro caso nos pareció más adecuado.
El MAS asociado a LES debe considerarse una complicación severa que pone en riesgo la vida del paciente, ya que su mortalidad es del 11% y el número de pacientes que requieren de admisión a unidad de cuidados intensivos pediatricos, ventilación mecánica y disfunción cardiovascular (el 63, el 53 y el 47%, respectivamente) es elevado3,10. La revisión de los 10 casos mostró una mortalidad del 10%, lo que concuerda con lo reportado. Nuestra paciente tuvo una evolución favorable y actualmente tiene 10 años de edad y lleva una vida normal.
ConclusiónEl MAS es una complicación poco frecuente de LES pero potencialmente fatal, por lo que su diagnóstico y tratamiento tempranos son fundamentales para mejorar la sobrevida. Se debe sospechar MAS en los pacientes con altos índices de actividad de LES. La CFM en nuestro caso fue eficaz para el tratamiento de las manifestaciones clínicas. Se debe de considerar su uso en pacientes con MAS asociado a LES a pesar de las citopenias profundas sin temor a empeorar el cuadro. La evidencia en cuanto al mejor tratamiento es escasa, dada la rareza de la entidad, por lo que es necesario individualizar los casos para determinar la terapia adecuada en estos pacientes.
Responsabilidades éticasProtección de personas y animalesLos autores declaran que para esta investigación no se han realizado experimentos en seres humanos ni en animales.
Confidencialidad de los datosLos autores declaran que han seguido los protocolos de su centro de trabajo sobre la publicación de datos de pacientes y que todos los pacientes incluidos en el estudio han recibido información suficiente y han dado su consentimiento informado por escrito para participar en dicho estudio.
Derecho a la privacidad y consentimiento informadoLos autores han obtenido el consentimiento informado de los pacientes y/o sujetos referidos en el artículo. Este documento obra en poder del autor de correspondencia.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.
A la unidad de investigación del Colegio Mexicano de Reumatología y al Dr. Luis Javier Jara Quezada, coordinador de la unidad, por su apoyo en el taller «Cómo escribir un artículo científico paso por paso».


