


El síndrome de VEXAS es una entidad infrecuente secundaria a una mutación en el gen UBA1, localizado en el cromosoma X. Esto provoca como consecuencia la aparición de vacuolas, característica de esta enfermedad, en células madre hematopoyéticas. Se caracteriza por la aparición de múltiples manifestaciones autoinflamatorias sistémicas y hematológicas, que responden y acaban siendo dependientes al tratamiento con glucocorticoides. En la presente publicación aportamos una serie de 2 casos diagnosticados en nuestro centro, y realizamos una breve revisión de la evidencia publicada al respecto.
VEXAS syndrome is a rare entity secondary to UBA1 gene mutations, located on the X chromosome. This mutation generates, as a consequence, a characteristic vacuolation on haematopoietic stem-cells. It is characterized by multiple autoinflammatory and haematologic manifestations, which respond and end up being dependent on corticosteroid treatment. In this publication we present a 2-case series diagnosed at our hospital and make a brief literature review of the published evidence so far.
El síndrome de VEXAS es una entidad incluida dentro de las enfermedades autoinflamatorias, causada por mutaciones en el gen UBA1, y descrita por primera vez por Beck et al. en el año 20201. El término VEXAS es un acrónimo formado por las siglas de las características fundamentales de la enfermedad1,2:
- -
Vacuolas: presencia de vacuolas citoplásmicas en células madre hematopoyéticas.
- -
Enzima E1 ubiquitina-ligasa: se produce una mutación en el gen UBA1, que codifica dicha enzima, activadora de la ubiquitinación.
- -
Cromosoma X: donde se encuentra el gen UBA1.
- -
Autoinflamatoria.
- -
Somática: la mutación es adquirida y no heredable, presente en las células madre hematopoyéticas, y ausente en otras células.
Las manifestaciones clínicas de esta entidad son muy variadas, siendo las más frecuentes las afectaciones hematológica, cutánea, reumatológica y pulmonar2. Además, es característica la corticodependencia, con la aparición de nuevos brotes al bajar la dosis de glucocorticoides (Gc), siendo muy difícil conseguir su retirada3. A continuación, aportamos una serie de 2 casos reseñables diagnosticados en nuestro centro, y realizamos una breve revisión al respecto.
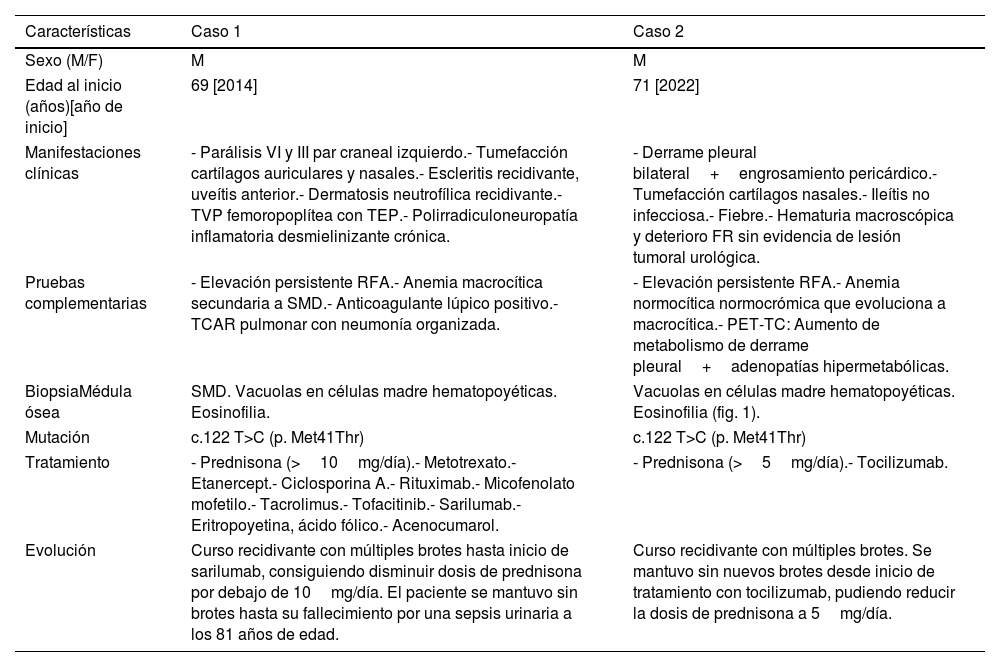
Observación clínicaEn la tabla 1 resumimos los datos clínicos relevantes de cada caso, así como los tratamientos recibidos y la evolución de los mismos. Como datos reseñables, ambos pacientes comenzaron alrededor de los 70 años, desarrollando clínica corticodependiente, especialmente condritis nasal y anemia macrocítica, y alcanzaron estabilidad clínica con el empleo de anti-interleucina 6. En el caso 1 predominó la clínica ocular y cutánea, junto con la aparición de fenómenos trombóticos, mientras que en el caso 2 presentó manifestaciones pulmonares, renales y gastrointestinales.
Descripción de los casos clínicos del síndrome de VEXAS diagnosticados en nuestro hospital
| Características | Caso 1 | Caso 2 |
|---|---|---|
| Sexo (M/F) | M | M |
| Edad al inicio (años)[año de inicio] | 69 [2014] | 71 [2022] |
| Manifestaciones clínicas | - Parálisis VI y III par craneal izquierdo.- Tumefacción cartílagos auriculares y nasales.- Escleritis recidivante, uveítis anterior.- Dermatosis neutrofílica recidivante.- TVP femoropoplítea con TEP.- Polirradiculoneuropatía inflamatoria desmielinizante crónica. | - Derrame pleural bilateral+engrosamiento pericárdico.- Tumefacción cartílagos nasales.- Ileítis no infecciosa.- Fiebre.- Hematuria macroscópica y deterioro FR sin evidencia de lesión tumoral urológica. |
| Pruebas complementarias | - Elevación persistente RFA.- Anemia macrocítica secundaria a SMD.- Anticoagulante lúpico positivo.- TCAR pulmonar con neumonía organizada. | - Elevación persistente RFA.- Anemia normocítica normocrómica que evoluciona a macrocítica.- PET-TC: Aumento de metabolismo de derrame pleural+adenopatías hipermetabólicas. |
| BiopsiaMédula ósea | SMD. Vacuolas en células madre hematopoyéticas. Eosinofilia. | Vacuolas en células madre hematopoyéticas. Eosinofilia (fig. 1). |
| Mutación | c.122 T>C (p. Met41Thr) | c.122 T>C (p. Met41Thr) |
| Tratamiento | - Prednisona (>10mg/día).- Metotrexato.- Etanercept.- Ciclosporina A.- Rituximab.- Micofenolato mofetilo.- Tacrolimus.- Tofacitinib.- Sarilumab.- Eritropoyetina, ácido fólico.- Acenocumarol. | - Prednisona (>5mg/día).- Tocilizumab. |
| Evolución | Curso recidivante con múltiples brotes hasta inicio de sarilumab, consiguiendo disminuir dosis de prednisona por debajo de 10mg/día. El paciente se mantuvo sin brotes hasta su fallecimiento por una sepsis urinaria a los 81 años de edad. | Curso recidivante con múltiples brotes. Se mantuvo sin nuevos brotes desde inicio de tratamiento con tocilizumab, pudiendo reducir la dosis de prednisona a 5mg/día. |
En la tabla encontramos sexo, edad, manifestaciones clínicas, principales alteraciones objetivadas en las pruebas complementarias, alteraciones presentadas en la biopsia de médula ósea, mutación diagnosticada, tratamientos recibidos y evolución clínica presentada.
F: femenino; FR: función renal; M: masculino; RFA: reactantes de fase aguda; SMD: síndrome mielodisplásico; TEP: tromboembolismo pulmonar; TVP: trombosis venosa profunda.
El síndrome de VEXAS es una enfermedad autoinflamatoria debida a una mutación adquirida en las células madre hematopoyéticas de la médula ósea, en el codón p.Met41 del gen que codifica la enzima E1 ubiquitina ligasa (UBA1), localizado en el brazo corto del cromosoma X (Xp11.23)4. Existen 3 mutaciones en UBA1 reconocidas como patogénicas para el síndrome de VEXAS (p.Met41Thr, p.Met41Leu y p.Met41Val), siendo la última la asociada a mayor gravedad clínica y mortalidad2,5. Recientemente, se han identificado otras mutaciones en localizaciones atípicas del mismo gen6. Todos los progenitores hematopoyéticos presentan dichas mutaciones, especialmente en células de estirpe mieloide (neutrófilos y monocitos), estando ausentes en linfocitos maduros1. Este mosaicismo estaría justificado porque la aparición de la mutación en las células de la estirpe linfoide facilita su apoptosis y resulta incompatible con su supervivencia celular6, haciendo que los pacientes presenten linfopenia, y los pocos linfocitos circulantes supervivientes no presenten mutación para el gen UBA1.
Respecto a su epidemiología, al igual que en nuestra serie, el paciente tipo con síndrome de VEXAS suele ser un varón, mayor de 50 años, de raza blanca2,3, describiéndose casos excepcionales en las mujeres. Se hipotetiza que esto es debido a que la expresión del gen UBA1 escapa a la inactivación del segundo cromosoma X en las mujeres, por lo que resulta más complicado que una mujer con mutación en el cromosoma no inactivado pueda desarrollar la enfermedad7.
Su patogenia todavía no es del todo bien conocida. Se relaciona con la presencia de la mutación en los precursores hematopoyéticos y sus células derivadas, especialmente neutrófilos y monocitos2. Esto provocaría la sobreexpresión de diversas citoquinas, precipitaría una activación proinflamatoria neutrofílica incontrolada y originaría el cuadro clínico8, ocasionando también un cambio en el microambiente de la médula ósea, facilitando la aparición de las alteraciones hematológicas descritas2.
En cuanto a la clínica del síndrome de VEXAS, es muy variada y afecta a diferentes sistemas del organismo4,7. Al igual que en nuestra serie, es característico que dichas manifestaciones sean corticodependientes, respondiendo rápidamente a dosis altas de Gc y recayendo con la bajada de dosis3,4. A nivel hematológico, la anemia macrocítica suele presentarse prácticamente en todos los pacientes2, al igual que vemos en nuestra serie, así como otras citopenias asociadas. Aproximadamente la mitad de los pacientes terminan desarrollando un síndrome mielodisplásico (SMD), como en uno de nuestros casos, que por lo general suele presentar fenotipos de bajo riesgo; así como episodios trombóticos (especialmente trombosis venosa profunda), consecuencia de la inflamación crónica y la disfunción endotelial ocasionadas9. A nivel cutáneo, las lesiones más frecuentes son las de dermatosis neutrofílica, como en el caso 1 de nuestra serie, la vasculitis cutánea y la paniculitis septal8. A nivel reumatológico, los pacientes suelen desarrollar cuadros de fiebre recurrente, síntomas constitucionales y linfadenopatías. Resulta bastante frecuente la aparición de policondritis recidivante, tal y como observamos en nuestra serie, con afección de cartílagos nasales y auriculares2. A nivel pulmonar, puede aparecer tos y disnea, simulando una neumonía o una descompensación de insuficiencia cardiaca resistentes a tratamiento con diuréticos o antibioterapia2, siendo frecuente encontrarnos infiltrados pulmonares intersticiales, consolidaciones o derrame pleural (como en nuestro caso 2), entre otros. A nivel ocular, lo más frecuente es la aparición de epiescleritis, escleritis y uveítis2, como podemos ver en el caso 1 de nuestra serie. Además, se ha descrito pericarditis, alteraciones del sistema nervioso periférico, como en nuestro caso 1, e incluso alteraciones gastrointestinales2 o alteraciones renales (hematuria y proteinuria), como en nuestro caso 2, entre otras.
Para su diagnóstico es fundamental la realización del test genético en aquellos pacientes con clínica compatible8. También es característica, aunque no patognomónica, la visualización de vacuolización citoplasmática de los precursores hematopoyéticos en el aspirado y la biopsia de médula ósea (fig. 1), especialmente en precursores mieloides y eritroides1. Cabe recordar que también es posible encontrar vacuolas intracitoplasmáticas en la intoxicación aguda alcohólica, sepsis, intoxicación por zinc, mieloma múltiple, déficit de cobre o, incluso, en los mismos SMD no asociados al síndrome de VEXAS2,7,8.
 y a 100 aumentos (×100, derecha). En ella podemos observar la serie mieloide correctamente representada, sin detención madurativa ni aumento de células blásticas. Especialmente destaca la vacuolización citoplasmática en células mieloides, característica del síndrome VEXAS. En la muestra obtenida también observamos mínimos cambios displásicos en la serie eritroide, así como la presencia de marcada eosinofilia.")
Biopsia de médula ósea de nuestro paciente número 2. Muestra histológica teñida con tinción de Giemsa. Visión al microscopio óptico a 50 aumentos (×50, izquierda) y a 100 aumentos (×100, derecha). En ella podemos observar la serie mieloide correctamente representada, sin detención madurativa ni aumento de células blásticas. Especialmente destaca la vacuolización citoplasmática en células mieloides, característica del síndrome VEXAS. En la muestra obtenida también observamos mínimos cambios displásicos en la serie eritroide, así como la presencia de marcada eosinofilia.
Todavía no existe un tratamiento estandarizado para el síndrome de VEXAS. La mayoría de recomendaciones terapéuticas están basadas en la experiencia clínica y en la evidencia extraída de escasos estudios retrospectivos2. Los objetivos fundamentales a conseguir con el tratamiento son el control de la inflamación, prevenir complicaciones asociadas (infecciones, citopenias y eventos trombóticos) y eliminar los clones hematopoyéticos con el gen UBA1 mutado.
A la hora de controlar la inflamación, como hemos visto en nuestra serie, los Gc a altas dosis son rápidamente eficaces (con un mínimo de 15-20mg/día de prednisona), permitiendo un control rápido de la sintomatología1,3,4. Sin embargo, sus efectos adversos hacen necesario el uso de otros fármacos para poder reducir su dosis. La respuesta a fármacos clásicos es muy limitada, siendo el metotrexato y la ciclosporina A los que más se han usado, con escaso éxito3,4,8. Se han obtenido mejores respuestas con diversos fármacos biológicos, especialmente con anti-interleucina 1 (como anakinra o canakinumab), anti-interleucina 6 (tocilizumab o sarilumab, como en nuestra serie) o inhibidores de JAK quinasas (parece ser que ruxolitinib es mejor que otros dentro de su grupo terapéutico, aunque el riesgo trombótico debería limitar su uso a no ser que ya estén anticoagulados, como ocurría en nuestro caso 1)2–4,8,10. Como estrategias para eliminar las células mutadas, se puede considerar el trasplante alogénico de progenitores hematopoyéticos en aquellos pacientes con peor pronóstico, dejando el tratamiento médico con inhibidores de la DNA metiltransferasa o inhibidores de JAK para aquellos pacientes de mejor pronóstico o que presentan contraindicaciones al trasplante2,3. En cuanto a la prevención de complicaciones asociadas, se ha recomendado tanto la profilaxis infecciosa como la vacunación en estos pacientes2–4. Para controlar las citopenias se pueden emplear tanto trasfusiones como agentes estimulantes de la citopoyesis3. Por último, se recomienda la antiagregación indefinida en pacientes que han tenido eventos trombóticos. La profilaxis antitrombótica primaria todavía se encuentra en debate, dada la ausencia de datos al respecto2,3.
ConclusiónDe reciente identificación en el año 2020, el síndrome de VEXAS es una enfermedad todavía novedosa y probablemente infradiagnosticada. Aún queda un largo camino hasta conseguir caracterizar por completo la enfermedad y encontrar terapias dirigidas efectivas que permitan controlar todas las manifestaciones autoinflamatorias de forma segura y efectiva.
Conflicto de interesesLos autores declaran que no presentan ningún tipo de conflicto de intereses para la realización y la publicación de este trabajo.


