

En el año 2015 la Sociedad Española de Reumatología (SER) publicó su posicionamiento sobre fármacos biosimilares. En esta actualización, la SER, sigue manifestando su compromiso inequívoco con la sostenibilidad del sistema sanitario de nuestro país y se alinea con las medidas que, sin reducir la calidad asistencial, estén encaminadas a asegurar su sostenibilidad. Desde la publicación del anterior posicionamiento la Comisión Europea ha autorizado la comercialización de nuevos fármacos biosimilares, lo que abre una excelente oportunidad de avanzar en la eficiencia de la atención sanitaria. En este nuevo escenario de incremento de la oferta terapéutica de biológicos, la SER considera imprescindible preservar la libertad de prescripción de los médicos que realizan la indicación de fármacos basándose exclusivamente en las características y circunstancias individuales de cada paciente, sin olvidar los aspectos económicos que se derivan de dicha actuación.
In 2015 the Spanish Society of Rheumatology (Sociedad Española de Reumatología [SER]) published its position paper on biosimilar drugs. In this update, the SER, continues to manifest its unequivocal commitment to the sustainability of the health system of our country and is aligned with the measures that, without reducing quality of care, are aimed at ensuring its continuity. Since the publication of the previous position paper, the European Commission has authorized new biosimilar drugs, which provides an excellent opportunity to advance the efficiency of health care. In this new scenario of increased therapeutic offer of biologics, the SER considers it crucial to preserve the freedom of prescription of physicians who prescribe drugs based exclusively on the characteristics and individual circumstances of each patient, without forgetting the economic aspects there of.
La incorporación de los fármacos biológicos (FB) ha supuesto una transformación del tratamiento de las enfermedades inflamatorias crónicas. Su perfil de eficacia y seguridad ha permitido desarrollar una actividad normal a muchos pacientes que hasta su llegada veían reducida su calidad de vida. Los FB tienen un elevado impacto económico y su utilización ha significado un esfuerzo relevante para el Sistema Nacional de Salud español1.
La autorización de comercialización de la Comisión Europea tras la opinión positiva de la European Medicines Agency (EMA) de fármacos biosimilares (BS) de los FB originales ha abierto una oportunidad de avanzar en la eficiencia de la atención sanitaria. Según la EMA, un BS es un FB que contiene una versión de la sustancia activa de un producto biológico original ya autorizado (fármaco de referencia [FR])2. Para su aprobación, un BS debe demostrar en un ejercicio de comparabilidad directa que las diferencias con su original no tienen efecto sobre su seguridad y eficacia.
La incorporación a la práctica clínica habitual de los BS plantea nuevos retos, como la libertad de prescripción del médico al elegir qué diana bloquear y con qué producto, la idoneidad de la sustitución masiva por motivos económicos o de la intercambiabilidad decidida por el binomio paciente-reumatólogo, o la posibilidad, no evaluada formalmente en ensayos clínicos (EC), de cambiar de un BS a otro BS por decisiones de compra centralizada tomadas bajo criterios exclusivamente económicos.
En 2015, la Sociedad Española de Reumatología (SER) publicó su posicionamiento sobre el uso de BS en las enfermedades reumáticas3. En este artículo se actualiza este posicionamiento a la luz de los nuevos datos que han aparecido desde su publicación.
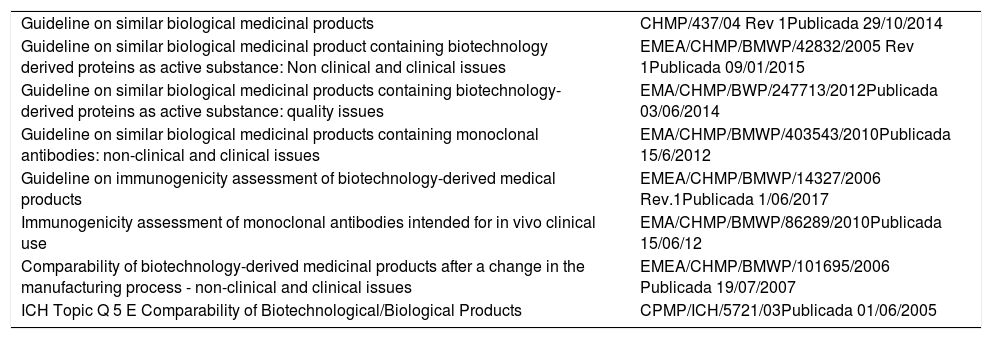
Normativa EMALa EMA, pionera en el desarrollo y establecimiento del proceso regulatorio para la autorización de BS, es la responsable de evaluar las solicitudes de los FB, incluidos BS, antes de su comercialización en la Unión Europea (UE). La EMA ha publicado distintas guías científicas sobre los BS (tabla 1). Según la legislación sobre medicamentos de la UE, los BS deben seguir un procedimiento de registro centralizado coordinado por EMA, y son evaluados por los mismos expertos que evalúan los FR. El Comité para Productos Médicos de Uso Humano (CHMP) emite una opinión científica sobre la aprobación y, finalmente, la Comisión Europea (CE) toma la decisión final sobre la autorización de comercialización en la UE. Este procedimiento centralizado es válido en todos los países de la UE, sin embargo, la EMA no realiza recomendaciones acerca de la intercambiabilidad entre FR y BS, y es cada estado el que decide sobre intercambiabilidad en base a la información científica disponible y a sus respectivos marcos legales. La EMA especifica que cualquier decisión sobre intercambio debe involucrar al prescriptor y al paciente4.
Principales guías EMA sobre biosimilares
| Guideline on similar biological medicinal products | CHMP/437/04 Rev 1Publicada 29/10/2014 |
| Guideline on similar biological medicinal product containing biotechnology derived proteins as active substance: Non clinical and clinical issues | EMEA/CHMP/BMWP/42832/2005 Rev 1Publicada 09/01/2015 |
| Guideline on similar biological medicinal products containing biotechnology-derived proteins as active substance: quality issues | EMA/CHMP/BWP/247713/2012Publicada 03/06/2014 |
| Guideline on similar biological medicinal products containing monoclonal antibodies: non-clinical and clinical issues | EMA/CHMP/BMWP/403543/2010Publicada 15/6/2012 |
| Guideline on immunogenicity assessment of biotechnology-derived medical products | EMEA/CHMP/BMWP/14327/2006 Rev.1Publicada 1/06/2017 |
| Immunogenicity assessment of monoclonal antibodies intended for in vivo clinical use | EMA/CHMP/BMWP/86289/2010Publicada 15/06/12 |
| Comparability of biotechnology-derived medicinal products after a change in the manufacturing process - non-clinical and clinical issues | EMEA/CHMP/BMWP/101695/2006 Publicada 19/07/2007 |
| ICH Topic Q 5 E Comparability of Biotechnological/Biological Products | CPMP/ICH/5721/03Publicada 01/06/2005 |
El uso de BS suscita controversias, especialmente en lo referente a intercambiabilidad y sustitución, conceptos que parecen próximos pero que desde el punto de vista técnico y médico-legal son diferentes, y a la incidencia que sobre la libertad de prescripción puedan tener criterios economicistas de los gestores.
En la legislación española, a la espera de una regulación específica sobre BS, la norma básica a seguir está contenida en la Ley 29/2006, de 26 de julio, de garantías y uso racional de los medicamentos y productos sanitarios, que interpreta la sustitución como el acto por el cual el farmacéutico puede, excepcionalmente y por motivos concretos como urgencia o desabastecimiento, dispensar un medicamento diferente al prescrito por el médico, sustituyéndolo por otro de igual composición, forma farmacéutica, vía de administración y dosificación, quedando excluidos de esa posibilidad los medicamentos que, por razón de sus características de biodisponibilidad y estrecho rango terapéutico, determine el Ministerio de Sanidad y Consumo. Así pues, la posibilidad de sustitución por parte del farmacéutico queda limitada a productos idénticos entre sí, lo que no es aplicable a los FB y sus BS. En la Orden SCO 2874-2007 de 28 de septiembre, se publicó la lista de medicamentos no sustituibles, que incluye a los FB, y en la Nota Informativa de la Agencia Española de Medicamentos y Productos Sanitarios de 24/4/2009, se hace expresa mención a ello.
En cuanto a la intercambiabilidad, el artículo 89.5 de la Ley de Garantías (Real Decreto Legislativo 1/2015, de 24 de julio) indica que «en el caso de los medicamentos BS, se respetarán las normas vigentes según regulación específica en materia de sustitución e intercambiabilidad», lo que indica que los BS tienen un régimen jurídico diferencial, pero no se concreta su regulación específica. Existe controversia sobre si solo se aplica a las oficinas de farmacia y no al hospital. Cabe recordar que actualmente hay BS, como los de insulina, comercializados en oficinas de farmacia, por lo que sería un contrasentido que no se permitiese la sustitución en oficinas de farmacia y sí en el ámbito hospitalario.
El Gobierno de España, en respuesta a una pregunta parlamentaria, ha reconocido que «los medicamentos biosimilares son FB y que como tal no son sustituibles o intercambiables en la dispensación sin el conocimiento del médico prescriptor»(BOCG, serie D n.o472, 5 junio de 2014, página 345).
Según nuestra legislación, queda claro que los FB no pueden ser sustituidos sin contar con el prescriptor, por lo que cualquier cambio entre FB tendrá que realizarse por iniciativa o de acuerdo con el prescriptor, de manera que ya no se tratará legalmente de una sustitución sino de un intercambio.
Dejando aparte la normativa legal, entre las sociedades científicas existe pleno acuerdo en cuanto a la necesidad de participación del clínico en la decisión de intercambiabilidad, expresado en sus respectivos posicionamientos. Únicamente la Sociedad Española de Farmacia Hospitalaria hace una matización atribuyendo a las Comisiones de Farmacia y Terapéutica la capacidad de decidir acerca del intercambio en el ámbito hospitalario dado que en ella están representados los prescriptores. Esta posibilidad de decisión colegiada no es aceptable, dado que la normativa que regula el funcionamiento de dicha comisión (Real Decreto 521/1987, de 15 de abril) no le atribuye esa competencia y que la responsabilidad del tratamiento no es colegiada sino personal y corresponde al médico, que es el depositario exclusivo de la competencia para prescribir, según el artículo 79 de la Ley de Garantías.
Información al paciente y libertad de prescripciónLa relación médico-paciente se basa en la confianza, la comunicación y la autonomía del paciente, por lo que este ha de estar informado del tratamiento que se le ofrece, y de acuerdo con él. Es una exigencia social, deontológica y legal, dado que la Ley 41/2002, de 14 de noviembre, básica reguladora de la autonomía del paciente y de derechos y obligaciones en materia de información y documentación clínica, indica en su artículo 2 que «toda actuación en el ámbito de la sanidad requiere, con carácter general, el previo consentimiento de los pacientes o usuarios», por lo que cualquier variación en las condiciones del paciente debe serle comunicada, incluyendo la necesidad de informar del cambio de un tratamiento concreto, debiendo ponderar el médico responsable la necesidad de un consentimiento por escrito, que será más necesario cuanto más dudoso sea el resultado de su intervención, como especifica el artículo 10 de dicha Ley.
Así pues en España no es legalmente posible la sustitución de biológicos a nivel de Farmacia Hospitalaria (FH) y la intercambiabilidad es responsabilidad del médico prescriptor que deberá tener en cuenta consideraciones éticas, clínicas y siempre con el consentimiento del paciente.
Dentro de la máxima colaboración con los gestores y FH, los médicos tenemos el derecho a seleccionar entre los medicamentos incluidos en el régimen de financiación estatal el que consideremos más apropiado para el paciente. Dicha capacidad está reconocida el artículo 36 de la Constitución, el artículo 4.7 de la Ley de Ordenación de las Profesiones Sanitarias, el artículo 77.1 de la Ley 29/06 y el Decreto 2065/1974.
Farmacovigilancia. Prescripción por nombre comercialCon la finalidad de asegurar la trazabilidad, en concreto en la atribución de potenciales efectos adversos, los FB se deben prescribir por nombre comercial. Según el artículo 5.12 del Real Decreto 577-2013 de 26 de julio, por el que se regula la farmacovigilancia de medicamentos de uso humano, se deben implementar medidas encaminadas a identificar el nombre del medicamento y número de lote en aquellas notificaciones que involucren a medicamentos de origen biológico o biotecnológico.
Controversias biosimilaresComplejidad estructural y fabricación de fármacos biológicosLos productos biológicos, incluyendo los BS, son proteínas grandes y complejas cuya producción requiere la maquinaria de transcripción y traslación de un organismo vivo, generalmente una célula de mamífero. Los FB pueden tener múltiples isoformas que difieren entre sí de forma mínima (microheterogeneidad) debido a: 1) modificaciones postraslacionales y 2) cambios menores en su proceso de fabricación, como diferentes medios de cultivo, de temperatura o técnicas de purificación5. Esta microheterogeneidad puede influir en la inmunogenicidad, hecho más relacionado con la eficacia clínica que con la seguridad.
Para producir un BS, se debe analizar exhaustivamente el FR y mediante ingeniería reversa6 determinar su secuencia de aminoácidos. Los desarrolladores de BS deben tener conocimiento y experiencia en el desarrollo y fabricación de FB, y deben establecer sus propios protocolos ya que los procesos de fabricación de los FR están protegidos por leyes de propiedad intelectual. Incluso pequeños cambios del proceso de fabricación pueden dar lugar a diferencias entre BS y FR respecto al nivel y naturaleza de la glucosilación, plegamiento proteico o capacidad de interaccionar con otras proteínas. Para recibir la aprobación regulatoria, estas modificaciones no deben afectar a la calidad, pureza o potencia del BS, ni dar lugar a cambios clínicamente significativos en seguridad o eficacia respecto al FR7.
Para crear un BS se utilizan herramientas y métodos analíticos sofisticados para demostrar que BS y FR no presentan diferencias relevantes estructuralmente. Sin embargo, un paquete de datos analíticos comparativos entre BS y FR no es suficiente para la aprobación del BS si no se aportan datos preclínicos (en animales) y clínicos (en humanos). Todos estos datos comprenden la «totalidad de la evidencia» con la que se evalúa el nivel de similitud del BS con su FR7–9.
Para su aprobación un BS debe pasar un largo camino de desarrollo que se puede dividir en 4 fases:
- 1.
Transfectar la línea celular elegida con el ADN que lleva las «instrucciones genéticas» para producir la secuencia de aminoácidos del biológico.
- 2.
En el proceso anterior se generan múltiples versiones del biológico, cada una producida por un «clon celular». Los científicos las evalúan para seleccionar el clon que produce el BS más parecido al FR.
- 3.
El proceso de fabricación se desarrolla y perfecciona a escala industrial para generar una cantidad del BS suficiente para su comercialización.
- 4.
Después del desarrollo del proceso de fabricación, se realizan pruebas preclínicas y clínicas. Las pruebas clínicas se someten a un proceso acelerado de evaluación clínica que a menudo implican 2 etapas en las que se compara el FR con el BS:
- •
Una primera con voluntarios sanos (Fase Ia) o pacientes (Fase Ib) para probar que el cuerpo humano procesa el BS de la misma manera que el FR, es decir su objetivo principal es la farmacocinética y la farmacodinámica.
- •
Una segunda etapa en un conjunto amplio de pacientes para probar que el BS tiene un nivel similar de eficacia, seguridad e inmunogenicidad que el FR.
La extrapolación de datos de eficacia y seguridad de un BS en una patología al resto de las indicaciones del FR es un concepto clave en las directrices de las agencias reguladoras y debe justificarse científicamente7,10,11. Esta debe basarse en la «totalidad de la evidencia», precisando datos no clínicos, análisis fisicoquímicos y funcionales que la justifiquen7,10–12. El proceso de comparación para establecer biosimilaridad se realiza bajo un enfoque paso a paso y a través de un exhaustivo ejercicio de comparabilidad, en el que cualquier diferencia relevante observada debe ser justificada. Inicialmente deben realizarse estudios no clínicos cuyo primer paso son los estudios in vitro, que deben ser lo suficientemente sensibles para detectar cualquier diferencia en la actividad biológica del BS y el producto biológico de referencia. El segundo paso consiste en valorar la necesidad de llevar a cabo estudios in vivo, donde debe evaluarse si, por ejemplo, existen efectos mediados por anticuerpos monoclonales no detectables en estudios in vitro. Sin embargo, pueden no ser necesarios si la comparabilidad de los estudios in vitro ha sido satisfactoria. El tercer paso sería la realización de estudios in vivo propiamente dichos, cuyo enfoque dependerá de la necesidad de información adicional, no siendo precisos estudios de toxicidad en humanos3. Sin embargo, cuando el FR tiene mecanismos de acción diferentes dependiendo de las enfermedades en las que esté indicado7,10,11, puede ser necesario realizar nuevos estudios funcionales y/o farmacodinámicos para la aprobación regulatoria mediante extrapolación. Estos datos adicionales proporcionan una mayor seguridad de que el BS tendrá una eficacia y seguridad clínica similar en las indicaciones extrapoladas a las que se han obtenido en la indicación en la que se ha realizado el EC12.
No se puede exigir más predictibilidad en términos de seguridad y eficacia a un BS que a cualquier otro producto nuevo que llega al mercado13. De hecho, el extenso análisis preclínico que se exige a los BS, incluyendo los que se evalúan para la concesión de la extrapolación, en realidad reducen el grado de incertidumbre de estos compuestos en comparación con cualquier otro nuevo producto.
Las agencias reguladoras no tienen un enfoque único respecto a la extrapolación de indicaciones. Estudian caso por caso y pueden llegar a diferentes decisiones. Por ejemplo, el infliximab original tiene aprobación para artritis reumatoide (AR), espondilitis anquilosante (EA), psoriasis, artritis psoriásica (APs), colitis ulcerosa (CU) y enfermedad de Crohn (EC)14. Los BS de infliximab, CT-P13 y SB2 solo han sido estudiados mediante ensayos de comparación directa con el innovador en pacientes con AR15,16 y EA17. La EMA, FDA y los reguladores coreanos aprobaron CT-P13 (Remsima® e Inflectra®) para todas las indicaciones del FR18,19. Sin embargo, Japón los aprobó solo para AR, EC y CU20 y la agencia canadiense no apoyó inicialmente la extrapolación de datos clínicos para EC o CU, exigiendo una evaluación clínica en estas patologías21,22. Recientemente, la agencia canadiense aprobó las indicaciones para EC, EC fistulizante y CU para este BS de infliximab basándose en la evidencias acumuladas sobre su similitud con el innovador respecto a calidad, mecanismo de acción, fisiopatología de la enfermedad, perfil de seguridad, régimen de dosificación, y sobre la experiencia clínica con el innovador23.
Tal y como se ha comentado, la extrapolación es una extensión científica lógica del concepto de BS. Sin embargo, se han expresado opiniones en su contra24–26 centradas en la seguridad, sobre todo en la edad pediátrica27 y en la eficacia clínica en las enfermedades inflamatorias intestinales en las que el mecanismo de acción es diferente al de la AR28. La decisión inicial de EMA y FDA de aceptar la extrapolación del BS de infliximab a enfermedades inflamatorias intestinales en lo que respecta a eficacia, seguridad e inmunogenicidad está siendo refrendada por estudios observacionales29–31 y 2 recientes revisiones sistemáticas32,33 que concluyen que no existen diferencias en eficacia, seguridad e inmunogenicidad respecto al FR.
Intercambio/sustituciónUna de las cuestiones más controvertidas es si en pacientes con enfermedad estable o controlada se puede cambiar el FR por el BS sin perder eficacia ni aumentar efectos adversos. Dos informes de la UE, para profesionales y para pacientes evalúan este aspecto. En el de profesionales se indica: «Las decisiones sobre intercambiabilidad y/o sustitución dependen de las autoridades competentes de cada país y están fuera del ámbito de la EMA y del CHMP»34. El de pacientes indica: «Toda decisión de intercambio terapéutico (cambiar un medicamento por otro) debe ser tomada por su médico consultándole a usted y teniendo en cuenta cualquier posible práctica establecida sobre el uso de medicamentos biológicos de su país. Para cualquier pregunta relativa al cambio de un medicamento biológico por otro, los pacientes deben consultar a su médico, farmacéutico o enfermero especializado»35.
La EMA y la CE han publicado conjuntamente una guía para profesionales de la salud, donde se recoge que: «La EMA no regula la intercambiabilidad, el cambio ni la sustitución de un medicamento de referencia por su BS. Estos son competencia de los Estados Miembros de la UE»4.Es decir, la autorización de un BS no significa que sea considerado intercambiable con su FR. Por tanto, la intercambiabilidad (práctica médica de cambiar un medicamento por otro con el cual se espera lograr el mismo efecto, por iniciativa o con la aprobación del médico prescriptor) en contraposición a la sustitución (práctica de sustituir el medicamento prescrito por otro equivalente por parte del farmacéutico en el acto de la dispensación, sin previa consulta o sin conocimiento del médico prescriptor) debe ser una decisión clínica del médico, realizada de forma individual, basada en la evidencia científica y con conocimiento y consentimiento del paciente. Sin embargo, en la literatura médica anglosajona el término intercambio (switching) puede referirse a: 1) Cambiar un FR por otro, 2) Cambiar un FR por un BS, 3) Cambiar un BS por otro y 4) Medical switch vs Non-Medical switch. Esto introduce más variabilidad y controversia al interpretar los resultados y, desde un punto de vista clínico, cada una de estas acepciones del término intercambio puede tener distintas consecuencias.
Tras la aprobación por la EMA del primer BS de un anticuerpo monoclonal en septiembre de 2013, múltiples estudios de extensión de los EC de distintos BS han evaluado el cambio del FR al BS, aunque con distintos diseños36. La mayoría han evaluado la transición del FR al BS y una minoría han estudiado el cambio simple (BS a FR y FR a BS después de la fase ciega) o cambios múltiples entre BS y FR. Según estos autores, la mayoría de los estudios no son sensibles para excluir posibles riesgos asociados con la práctica de alternar entre FB que son muy similares (pero no idénticos) entre sí.
En la extensión a 102 semanas de CT-P13, los pacientes aleatorizados a Remicade® cambiaron a CT-P13, sin observarse diferencias en medidas de eficacia, efectos adversos ni en formación de anticuerpos antifármaco (AAF)37. En la extensión del PLANETAS tampoco se apreciaron diferencias en eficacia, seguridad e inmunogenicidad, aunque existía una disparidad numérica con un porcentaje mayor de efectos adversos y formación de AAF en el grupo con cambio del FR al BS: 4,8 vs. 3,3% y 27,4 vs. 23,3% respectivamente38. En la extensión del SB2 (Flixabi®), a 78 semanas, se realeatorizaron en la semana 54 a los pacientes del grupo inicial del Remicade® a seguir con él o cambiar a SB2, mientras que los pacientes aleatorizados inicialmente a SB2 continuaron con él. Un total de 94 cambiaron a SB2 y 101 continuaron con Remicade®; en la semana 78 el perfil de seguridad, eficacia e inmunogenicidad fue comparable entre los 3 grupos (SB2/SB2, Remicade®/SB2 y Remicade®/Remicade®)39.
En la fase de extensión del SB4, que incluyo a 254 pacientes, 126 continuaron con SB4 y 119 cambiaron de Enbrel® a SB4, observándose respuesta clínica, PROS y datos de progresión radiológica comparables40. En el estudio EGALITY se realizaron cambios múltiples entre Enbrel® y GP2015. El estudio tenía 4 periodos: a) cribado, b) periodo de tratamiento 1 (semana 0-12) aleatorización 1:1 a GP2015 (n=264) o Enbrel® (n=267), c) periodo tratamiento 2 (semana 13-30) en el que los pacientes con al menos una mejora del 50% en el PASI fueron aleatorizados a continuar con GP2015 o a una secuencia de tratamiento alternativo entre GP2015 y Enbrel® cada 6 semanas, y d) fase de extensión (semana 31-52) en la que los pacientes seguían recibiendo el tratamiento administrado durante las 6 semanas finales del periodo 2. En semana 30 y 52 no se observaron diferencias en la variable de desenlace primaria, PASI 75 ni resto de variables. También se observó que los cambios múltiples entre Enbrel® y GP2015 no incidían sobre eficacia, ni había diferencias en inmunogenicidad y seguridad41 Finalmente una revisión sobre BS de adalimumab no encontró diferencias en eficacia, seguridad o inmunogenicidad entre Humira® y sus BS42.
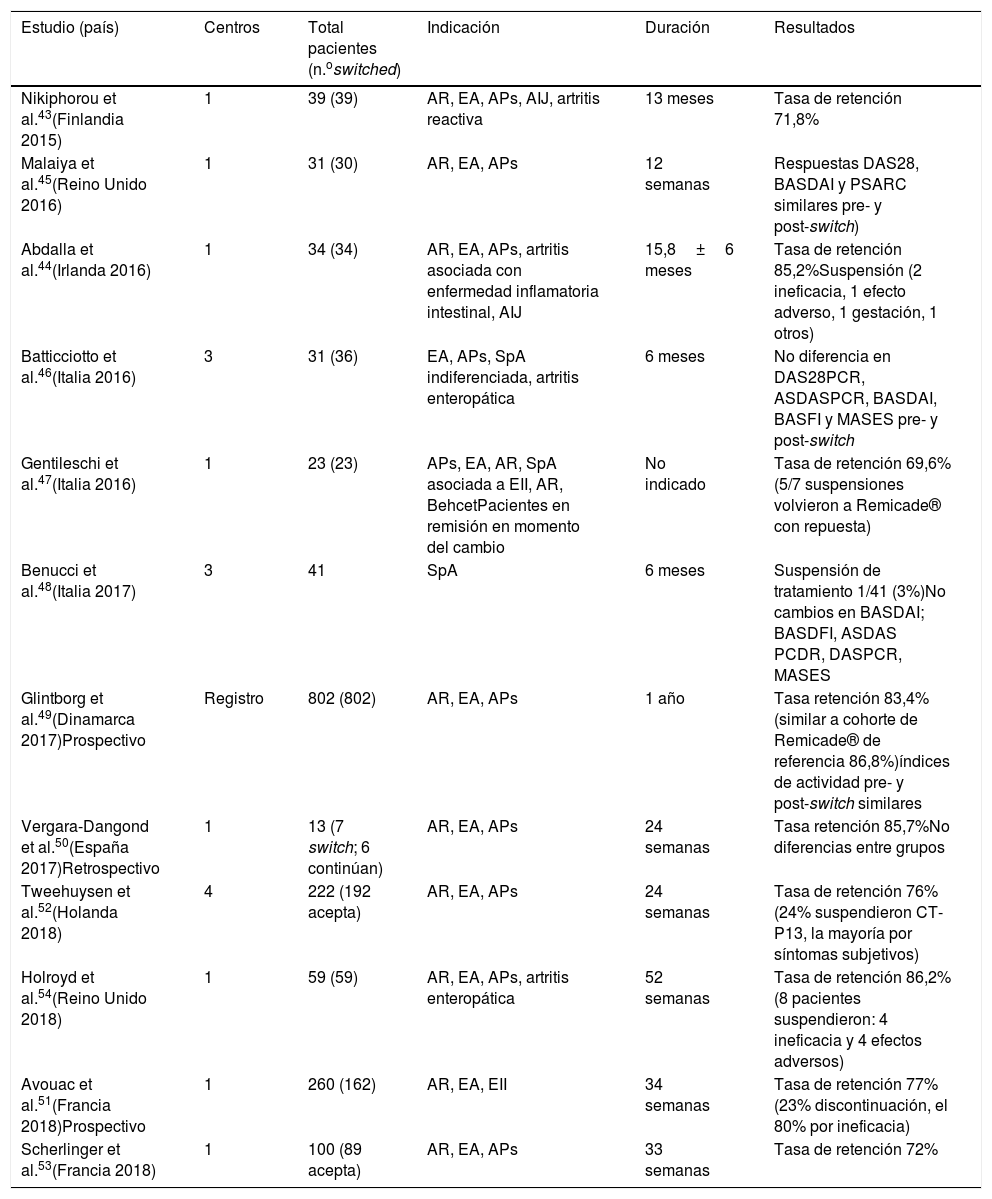
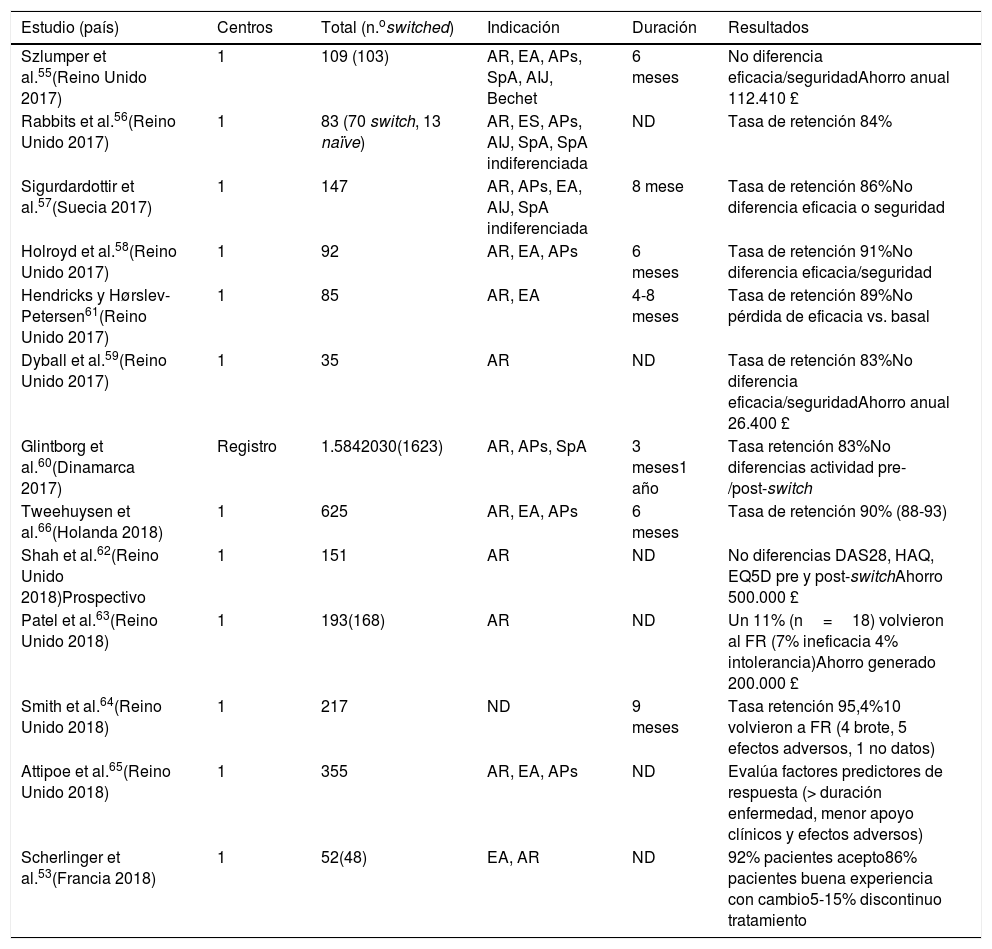
Se han publicado múltiples estudios de vida real con intercambio de Remicade® a CTP13 (tabla 2)43–54 y de Enbrel® a SB4 (tabla 3)55–66, sin observarse pérdidas de eficacia, problemas de seguridad ni cambios en perfil de inmunogenicidad; en algunos se evaluaban las tasas de persistencia del BS, y esta era similar a las de las cohortes de referencia. Solo en un estudio (23 pacientes) y donde el cambio se realizó por motivos regulatorios47 se observó una pérdida de eficacia; los autores concluyen que la sustitución no debe realizarse por motivos económicos o legales. No hacen referencia al posible papel del efecto nocebo67,68.
Estudios de vida real con intercambio de Remicade® a CTP13
| Estudio (país) | Centros | Total pacientes (n.oswitched) | Indicación | Duración | Resultados |
|---|---|---|---|---|---|
| Nikiphorou et al.43(Finlandia 2015) | 1 | 39 (39) | AR, EA, APs, AIJ, artritis reactiva | 13 meses | Tasa de retención 71,8% |
| Malaiya et al.45(Reino Unido 2016) | 1 | 31 (30) | AR, EA, APs | 12 semanas | Respuestas DAS28, BASDAI y PSARC similares pre- y post-switch) |
| Abdalla et al.44(Irlanda 2016) | 1 | 34 (34) | AR, EA, APs, artritis asociada con enfermedad inflamatoria intestinal, AIJ | 15,8±6 meses | Tasa de retención 85,2%Suspensión (2 ineficacia, 1 efecto adverso, 1 gestación, 1 otros) |
| Batticciotto et al.46(Italia 2016) | 3 | 31 (36) | EA, APs, SpA indiferenciada, artritis enteropática | 6 meses | No diferencia en DAS28PCR, ASDASPCR, BASDAI, BASFI y MASES pre- y post-switch |
| Gentileschi et al.47(Italia 2016) | 1 | 23 (23) | APs, EA, AR, SpA asociada a EII, AR, BehcetPacientes en remisión en momento del cambio | No indicado | Tasa de retención 69,6% (5/7 suspensiones volvieron a Remicade® con repuesta) |
| Benucci et al.48(Italia 2017) | 3 | 41 | SpA | 6 meses | Suspensión de tratamiento 1/41 (3%)No cambios en BASDAI; BASDFI, ASDAS PCDR, DASPCR, MASES |
| Glintborg et al.49(Dinamarca 2017)Prospectivo | Registro | 802 (802) | AR, EA, APs | 1 año | Tasa retención 83,4% (similar a cohorte de Remicade® de referencia 86,8%)índices de actividad pre- y post-switch similares |
| Vergara-Dangond et al.50(España 2017)Retrospectivo | 1 | 13 (7 switch; 6 continúan) | AR, EA, APs | 24 semanas | Tasa retención 85,7%No diferencias entre grupos |
| Tweehuysen et al.52(Holanda 2018) | 4 | 222 (192 acepta) | AR, EA, APs | 24 semanas | Tasa de retención 76% (24% suspendieron CT-P13, la mayoría por síntomas subjetivos) |
| Holroyd et al.54(Reino Unido 2018) | 1 | 59 (59) | AR, EA, APs, artritis enteropática | 52 semanas | Tasa de retención 86,2% (8 pacientes suspendieron: 4 ineficacia y 4 efectos adversos) |
| Avouac et al.51(Francia 2018)Prospectivo | 1 | 260 (162) | AR, EA, EII | 34 semanas | Tasa de retención 77% (23% discontinuación, el 80% por ineficacia) |
| Scherlinger et al.53(Francia 2018) | 1 | 100 (89 acepta) | AR, EA, APs | 33 semanas | Tasa de retención 72% |
AIJ: artritis idiopática juvenil; APs: artritis psoriásica; AR: artritis reumatoide; EA: espondilitis anquilosante; EII: enfermedad inflamatoria intestinal; SpA: espondiloartritis.
Estudios de vida real con intercambio de Enbrel® a SB4
| Estudio (país) | Centros | Total (n.oswitched) | Indicación | Duración | Resultados |
|---|---|---|---|---|---|
| Szlumper et al.55(Reino Unido 2017) | 1 | 109 (103) | AR, EA, APs, SpA, AIJ, Bechet | 6 meses | No diferencia eficacia/seguridadAhorro anual 112.410 £ |
| Rabbits et al.56(Reino Unido 2017) | 1 | 83 (70 switch, 13 naïve) | AR, ES, APs, AIJ, SpA, SpA indiferenciada | ND | Tasa de retención 84% |
| Sigurdardottir et al.57(Suecia 2017) | 1 | 147 | AR, APs, EA, AIJ, SpA indiferenciada | 8 mese | Tasa de retención 86%No diferencia eficacia o seguridad |
| Holroyd et al.58(Reino Unido 2017) | 1 | 92 | AR, EA, APs | 6 meses | Tasa de retención 91%No diferencia eficacia/seguridad |
| Hendricks y Hørslev-Petersen61(Reino Unido 2017) | 1 | 85 | AR, EA | 4-8 meses | Tasa de retención 89%No pérdida de eficacia vs. basal |
| Dyball et al.59(Reino Unido 2017) | 1 | 35 | AR | ND | Tasa de retención 83%No diferencia eficacia/seguridadAhorro anual 26.400 £ |
| Glintborg et al.60(Dinamarca 2017) | Registro | 1.5842030(1623) | AR, APs, SpA | 3 meses1 año | Tasa retención 83%No diferencias actividad pre-/post-switch |
| Tweehuysen et al.66(Holanda 2018) | 1 | 625 | AR, EA, APs | 6 meses | Tasa de retención 90% (88-93) |
| Shah et al.62(Reino Unido 2018)Prospectivo | 1 | 151 | AR | ND | No diferencias DAS28, HAQ, EQ5D pre y post-switchAhorro 500.000 £ |
| Patel et al.63(Reino Unido 2018) | 1 | 193(168) | AR | ND | Un 11% (n=18) volvieron al FR (7% ineficacia 4% intolerancia)Ahorro generado 200.000 £ |
| Smith et al.64(Reino Unido 2018) | 1 | 217 | ND | 9 meses | Tasa retención 95,4%10 volvieron a FR (4 brote, 5 efectos adversos, 1 no datos) |
| Attipoe et al.65(Reino Unido 2018) | 1 | 355 | AR, EA, APs | ND | Evalúa factores predictores de respuesta (> duración enfermedad, menor apoyo clínicos y efectos adversos) |
| Scherlinger et al.53(Francia 2018) | 1 | 52(48) | EA, AR | ND | 92% pacientes acepto86% pacientes buena experiencia con cambio5-15% discontinuo tratamiento |
AIJ: artritis idiopática juvenil; APs: artritis psoriásica; AR: artritis reumatoide; EA: espondilitis anquilosante; SpA: espondiloartritis.
De los estudios de vida real en los que se realizaba el cambio de Remicade® a CTP-13, el que más pacientes ha incluido es el de Glintborg et al.49, donde se realizó la sustitución de Remicade ® por CTP-13 de forma obligatoria. Se incluyó a 802 pacientes (403 AR, 279 EA y 120 APs) sin observarse cambios en las medidas de eficacia evaluadas 3 meses antes, en el momento del cambio y 3 meses tras cambio. Las tasas de retención fueron algo más bajas en la cohorte con CT-P13 (84,4%) versus la cohorte histórica con Remicade®(86,6%), con una diferencia de riesgo absoluto ajustada del 3,4% (p=0,03). Los autores indican que la diferencia no es necesariamente atribuible a CT-P13, sino que también podría representar un «efecto nocebo»67,68. Entre los estudios que realizaban el cambio de Enbrel® a SB4, el que mayor número de pacientes incluyó es el estudio BIO-SPAN, con un total de 642 pacientes (433 AR, 128 APs y 64 EA), donde se evaluó supervivencia y efectividad del cambio de Enbrel® a SB4, y usando como cohorte histórica comparativa a pacientes tratados con Enbrel® en 201466. La tasa de persistencia a los 6 meses de SB4 fue del 90% (IC 88-93%) versus la cohorte histórica de Enbrel® del 92% (IC 90-96%). Al comparar con la cohorte histórica, los pacientes que realizaron el cambio a SB4 tenían un riesgo relativo de discontinuación del tratamiento mayor: 1,57 (IC 95% 1,05-2,36). Estas diferencias son consideradas clínicamente irrelevantes y se sugieren 2 explicaciones: a) en la cohorte SB4 las suspensiones fueron más frecuentes por causas más subjetivas, por efecto nocebo67–69 y b) las pequeñas diferencias entre las cohortes podrían explicarse por un sesgo de tiempo del calendario, que relacionan con la mayor adherencia a las estrategias Treat-to-target en el año 2016 que en el 2014.
El estudio que parece confirmar la eficacia y seguridad del cambio desde el FR a su BS, es el NOR-SWITCH70, un ensayo de no inferioridad, de 52 semanas. El objetivo primario era evaluar si el cambio a CTP-13 no es inferior a Remicade®, con relación al no empeoramiento de la enfermedad, evaluándose asimismo, seguridad, inmunogenicidad y eficacia de CT-P13 versus Remicade®. Se incluyeron 482 pacientes (155 EC, 93 CU, 91 espondiloartritis, 77 AR, 30 APs y 30 psoriasis) en tratamiento estable con Remicade® durante al menos 6 meses y se aleatorizaron a seguir con Remicade® (n=241) o a cambiar a CTP-13 (n=241). Para definir empeoramiento se utilizaron medidas de desenlace estandarizadas, o consenso médico-paciente de empeoramiento de la enfermedad y de la necesidad de cambio de tratamiento. El margen de no inferioridad fue del 15%. Un 26% de los pacientes con Remicade® empeoro vs. 30% del grupo con CTP-13. La diferencia ajustada de tratamientos con un IC del 95% fue de −12,7% al 3,9%, dentro del margen de no inferioridad establecido, indicando que CTP-13 no es inferior al Remicade®. No hubo diferencias en efectos adversos ni variables de eficacia. Respecto a la inmunogenicidad, no se hallaron diferencias entre grupos en aparición de AAF durante cualquier momento del estudio, siendo del 11% en el grupo de Remicade® vs. del 13% en el grupo de CTP-13. El estudio no estaba diseñado para comparar las estrategias de tratamiento de forma individual en cada una de las enfermedades. Sin embargo, el resultado del estudio apoya la idea de que Remicade® puede ser intercambiado por su CTP-13 sin comprometer eficacia y seguridad.
En un reciente estudio se recogen las tasas de suspensión debido a efectos adversos y tasa total de suspensión en ensayos doble ciego de distintos BS, y no se observaban diferencias en las fases abiertas de estos ensayos en los que se hacia el switch del FR al BS69.
Los datos de los estudios NOR-SWITCH y DANBIO60,70 junto con actividades educativas y formativas dirigidas a médicos y pacientes son clave para la aceptación y uso de los BS. Hay ejemplos de que la formación e información conducen a implementar políticas de intercambiabilidad. En el Hospital de Southampton, digestólogos y reumatólogos desarrollaron un programa de ganancias compartidas para implantar el cambio del FR a CTP-1371,72. El programa se consensuó entre gestores, enfermeras, clínicos y pacientes. Estos acuerdos producen ahorro de costes y permiten invertir en servicios médicos, manteniendo los resultados clínicos71. Sin embargo, a pesar de la evidencia publicada, persisten dudas sobre aspectos como la generalización de datos de estos estudios a otros BS, o la eficacia y la seguridad cuando se produzca el intercambio múltiple entre distintos BS de un mismo FR36,73.
InmunogenicidadLa mayoría de los FB son capaces de inducir una respuesta inmune. La respuesta inmunológica es compleja y tanto el sistema inmune innato como el adquirido pueden contribuir al desarrollo de efectos adversos, pero es generalmente la formación de AAF lo que se conoce con el término de inmunogenicidad. Aunque la inmunogenicidad puede causar el desarrollo de reacciones inmunológicas agudas y potencialmente peligrosas74, los AAF no suelen tener repercusión clínica sobre la seguridad, mientras que su presencia se relaciona con la pérdida de eficacia parcial o total del fármaco.
Diversos factores influyen en la inmunogenicidad. Unos relacionados con el paciente, genética, edad, enfermedad de base, y tratamientos concomitantes, y otros con el producto dosis, vía de administración, procesos de manufacturado, formulación, estabilidad e impurezas75,76.
En el año 2015 la EMA actualizó las guías para la valoración de la inmunogenicidad de proteínas terapéuticas producidas por biotecnología (tabla 1). Esta guía recoge las estrategias y metodología para el estudio de la inmunogenicidad de FB y los requerimientos para la comparación de la inmunogenicidad de los BS frente a FR. Se especifica que los estudios comparativos son imprescindibles en el desarrollo de los BS y que todos los ensayos deben medir la inmunogenicidad tanto en el BS como en el FR. Se deben describir la incidencia y la naturaleza del desarrollo de estos anticuerpos en los que se incluyen la reactividad cruzada, los epítopos frente a los que se dirige y la actividad neutralizante o no. Aunque los BS están producidos por líneas celulares diferentes de los FR, no se han encontrado diferencias significativas en su inmunogenicidad en los EC pivotales, ni en los estudios de extensión de los distintos BS aprobados37,70. Sin embargo las agencias reguladoras especifican que es imprescindible la farmacovigilancia para detectar los eventos que puedan aparecer durante la comercialización (tabla 1).
La frecuencia de inmunogenicidad descrita en los ensayos de registro de los fármacos BS y los FR es habitualmente mayor y diferente de la que se ha descrito durante su uso en práctica clínica77. El motivo de esta discrepancia es el empleo de técnicas más sensibles y con menos interferencia del fármaco durante la práctica clínica. Esto supone un avance desde el punto de vista metodológico y científico, pero no significa que tenga ninguna repercusión clínica, por lo que la mera descripción de la frecuencia de aparición de AAF sin que se acompañe de su efecto en la farmacocinética de la proteína terapéutica es poco relevante.
El análisis de los estudios PLANETRA y PLANETAS describen diferencias en la producción de AAF entre pacientes con AR y EA, relacionados con la diferente dosis y tratamiento concomitante utilizado en el ensayo, pero sin diferencias entre el CTP-13 y Remicade®24. Dos grupos independientes han demostrado que los anticuerpos antiinfliximab son capaces de reconocer el CTP-13 y que se dirigen sobre todo hacia la región del anticuerpo relacionada con la unión al TNF y no se influye por los residuos glucosilados de la molécula de IgG78,79. Asimismo, mediante el estudio de los AAF aparecidos en los estudios PLANETRA y PLANETAS se demostró que tienen reacción cruzada, lo que significa que la respuesta inmunológica frente al Remicade® y al CTP-13 se dirige contra los mismos epítopos80. Un estudio recientemente publicado para determinar si los AAF frente a infliximab en pacientes con enfermedad inflamatoria intestinal reaccionaban de forma cruzada con Remicade®, CT-P13 y SB2, encontró que los AAF de pacientes tratados con CTP-13 o pacientes cambiados de Remicade® a CT-P13 muestran una reactividad cruzada completa con CT-P13 y SB2. Estos hallazgos indican que los epítopos inmunodominantes del FR y CT-P13 están igualmente presentes en SB281.
En el ensayo de fase iii que compara el SB4 con Enbrel®, se encontraron AAF al menos en una determinación en 2 pacientes (0,7%) en el grupo tratados con el SB4 y 39 (13,1%) en el grupo de Enbrel®. Los AAF aparecieron en ambos casos muy pronto, entre las semanas 2 y 8, y prácticamente todos desaparecieron en la semana 12, los AAF fueron transitorios y no neutralizantes82. La presencia de AAF no tuvo relevancia clínica y no afectó a los niveles del fármaco. Se han descrito resultados similares con GP201583.
La posición de los pacientes ante el uso de biosimilaresLos BS tienen el potencial de mejorar la atención del paciente, incrementando la eficiencia de los sistemas de salud, optimizando el acceso a los FB y ampliando las opciones de tratamiento disponibles. En consecuencia, la disponibilidad de BS podría conducir a un uso más generalizado de los FB, lo que podría redundar en mejores resultados en salud84.
Se han realizado encuestas sobre BS y FR a pacientes, grupos de pacientes, médicos y población general85–91, para obtener información básica sobre el uso y el conocimiento sobre los BS. En general los pacientes piensan que los FR son superiores en eficacia a los BS, sin percibir diferencias en la seguridad entre ambos85. Estos estudios muestran que la información proporcionada a los pacientes a través de sus médicos y asociaciones suele ser de mayor calidad, permitiéndoles asumir un rol más activo en la toma de decisiones terapéuticas, incluyendo el uso de BS.
En una encuesta realizada a pacientes y reumatólogos, el 49% de los pacientes sabía lo que era un BS, información recibida sobre todo a través de asociaciones de pacientes. Las diferencias percibidas entre FR y BS fueron mayores entre médicos que entre pacientes. Se observó que, con independencia del precio, los pacientes confiaban en la opinión del médico a la hora de optar por uno u otro, mientras que entre los reumatólogos, a igualdad de precio, la prescripción se decantaba por el original. Los aspectos que más condicionaban a los pacientes a la hora de cambiar un original por un BS eran: la opinión del médico y que el BS tuviera eficacia contrastada para su enfermedad. Por otra parte, un 28% de los reumatólogos encuestados creía que un BS y un original nunca deberían ser intercambiados, y un 39% no apoyaba la extrapolación de indicaciones92. En otra encuesta, la mayoría de los especialistas estaban dispuestos a empezar con un BS en un paciente naïve a biológicos, pero solo una minoría contemplaba el cambio de un original a un BS93.
Los BS son una realidad y es previsible su uso creciente. Por tanto debemos ser capaces de informar a nuestros pacientes sobre:
- -
¿Qué es un BS y qué exigen las agencias reguladoras para su aprobación?
- -
¿Cuál es su eficacia y seguridad?
- -
¿Qué pasa con el intercambio de un FR a un BS?
- -
¿Por qué un BS es más barato que el FR?
- -
¿Puede la FH cambiar de un FR al BS sin consentimiento de médico y paciente?
Es obvio que la consecución de este objetivo formativo debe contar con médicos, sociedades científicas, pacientes, asociaciones de enfermos, FH y gestores. Por tanto, la participación activa en el proceso de toma de decisiones terapéuticas por parte de un paciente formado e informado es inexcusable. El documento para pacientes recientemente publicado por la UE debería hacerse disponible a todos los pacientes que fueran a recibir estos medicamentos35.
Posicionamiento de la Sociedad Española de ReumatologíaDesde la SER manifestamos nuestro inequívoco compromiso con la sostenibilidad del sistema sanitario de nuestro país y nos alineamos con las medidas que, sin reducir la calidad asistencial, estén encaminadas a asegurar su continuidad. La autorización de comercialización por parte de EMA de BS de los biológicos originales va a abrir una excelente oportunidad de avanzar en la eficiencia de la atención sanitaria y va a mejorar el acceso de los pacientes reumáticos a las terapias biológicas.
En este nuevo escenario de incremento de la oferta terapéutica de biológicos, desde la SER consideramos imprescindible preservar la libertad de prescripción de los médicos que realizan la indicación de fármacos según las características y circunstancias individuales de cada paciente, sin olvidar los aspectos económicos que se derivan de dicha actuación.
Un BS es un FB que contiene una versión de la sustancia activa de un producto biológico original ya autorizado y para su aprobación debe demostrar que la variabilidad presente y cualquier diferencia respecto al FR no tiene efecto sobre su seguridad y eficacia. Por tanto, una vez que un BS ha sido autorizado, las agencias reguladoras garantizan que no existen diferencias significativas respecto al FR en calidad, eficacia y seguridad.
Por tanto, la SER quiere poner de manifiesto sobre los fármacos BS que:
- 1.
Un fármaco BS es un FB que ha demostrado biosimilitud en estudios in vitro con su fármaco de referencia, del que es indistinguible en términos de calidad, actividad biológica, seguridad y eficacia, en el marco de ensayos clínicos de comparación directa aleatorizados doble ciego.
- 2.
La elección de qué diana bloquear y con qué principio activo es responsabilidad del médico prescriptor y se debe decidir exclusivamente en el contexto de la relación médico-paciente, teniendo en cuenta las características de la enfermedad a tratar, las comorbilidades que puedan darse y tras informar al paciente.
- 3.
Una vez elegida qué diana bloquear y con qué principio activo, la elección de un fármaco innovador o un BS es responsabilidad del médico y se debe decidir exclusivamente en el contexto de la relación médico-paciente. En esta decisión se tendrán en cuenta la seguridad y coste/efectividad.
- 4.
El intercambio de un biológico por su BS debe ser realizado exclusivamente por el médico prescriptor, con el consentimiento del paciente. En el caso de pacientes con enfermedad estable puede ser aceptable un intercambio entre el FR y su BS, aunque debe ser una decisión individualizada y con el consentimiento del paciente.
- 5.
Actualmente no hay evidencias científicas sobre la eficacia y la seguridad del intercambio entre distintos BS de un mismo fármaco de referencia. Esto debería de ser tenido en cuenta e informar sobre este punto al paciente en el caso de que el médico prescriptor aconseje un intercambio entre BS.
- 6.
La SER entiende que las instituciones hospitalarias deben garantizar que todos los FB y BS que estén financiados por las autoridades sanitarias de nuestro país para el manejo de las enfermedades reumáticas estén disponibles en todos los hospitales del Sistema Nacional de Salud.
- 7.
Los fármacos BS están sujetos a una monitorización de seguridad igual a la de sus FR, por lo que es necesario favorecer su inclusión en los registros de farmacovigilancia específicos actualmente en desarrollo. La SER tiene amplia experiencia en estos registros y se ofrece para llevar a cabo estos estudios de seguridad.
- 8.
La trazabilidad de los FB es un elemento de calidad que permite asignar de forma específica a cada lote y producto las sospechas de reacciones adversas. Actualmente al BS se le asigna el mismo denominador común internacional que al innovador por lo que la prescripción debe de realizarse por marca comercial con la finalidad de conseguir una trazabilidad adecuada.
- 9.
En el caso de que el FR tenga más de una indicación, la extrapolación de indicaciones debe justificarse según los estándares de la EMA.
- 10.
El uso óptimo de los BS requiere diálogo e interacción continuos entre médicos, FH, asociaciones de pacientes y entidades reguladoras, con la intención de preservar el derecho a la salud de los pacientes y el objetivo de ofertarles productos de calidad, eficaces y seguros.
- 11.
Este posicionamiento de la SER se actualizará periódicamente a la luz de nuevas evidencias, estimándose la próxima dentro de 2 años.
M. Á. Abad Hernández. Ha colaborado y recibido honorarios de MSD, Pfizer, Celgene, Kern, Novartis, Biogen, Sandoz, Amgen y Janssen, como consultor, ponente, investigador y/o miembro de advisory boards.
J. L. Andreu. Honorarios por ponencias Abbvie, Antares, GSK, MSD, Nordic, Novartis, Sanofi, UCB. Honorarios por proyectos de investigación y consultorías: Abbvie, Amgen, AstraZeneca, Biogen, Cellgene, Celltrion, Fresenius Kabi, Gebro, Pfizer, Regeneron.
A. Balsa Criado. Ha recibido honorarios por presentaciones, asesorías o fondos para investigación de: Abbvie, MSD, Pfizer, UCB, Roche, BMS, Nordic, Sandoz, Lilly, Sanofi y Novartis.
F. Díaz-González. Por ponencias: Pfizer, MSD, Lilly, Janssen, BMS, Roche. Por asesoría científica: Lilly, Novartis, Pfizer, Amgen, Biogen, Celgene. Por proyecto de investigación: MSD, Abbvie, Roche, Novartis.
J. V. Moreno Muelas. Gebro, Janssen, MSD, Pfizer y Sanofi.
R. Queiro Silva. Ha colaborado y recibido honorarios de Abbvie, MSD, Pfizer, Celgene, UCB, Lilly, Novartis y Janssen como consultor, ponente, investigador y/o miembro de advisory boards.
J. J. Gómez-Reino. Advisory board y consultas: Abbvie, Biogen, BMS, Gebro, GSK, Lilly, Novartis, Pfizer, Roche, R-Pharma, Sandoz, Sanofi, Regeneron; Conferencias: Abbvie, BMS, Celgene, Janssen and Janssen, Lilly, MSD, Pfizer, Roche, Sanofi, UCB; becas: MSD, Pfizer, Roche, UCB.


