


Anticuerpos antifosfatidilserina/protrombina (aPS/PT) han sido descritos en poliarteritis nodosa (PAN) cutánea, en asociación con manifestaciones específicas.
ObjetivosDeterminar anticuerpos aPS/PT en pacientes con PAN y analizar su correlación con manifestaciones clínicas.
MétodosEstudio transversal comparativo de pacientes con PAN y 20 controles (10 con poliangitis microscópica [PAM] y 10 con enfermedad de Behçet [EB]). Se evaluaron variables demográficas, clínicas, serológicas y tratamiento; índices de pronóstico, actividad y daño. Se determinaron anticuerpos aPS/PT, anticardiolipina (aCL), anti-beta 2 glicoproteína 1 (anti-B2GP1) y anticoagulante lúpico (AL).
ResultadosFueron incluidos 14 pacientes con PAN, 11 (79%) mujeres, con duración de la enfermedad de 207 meses, y principalmente enfermedad inactiva. Sólo un paciente con PAN y uno con EB fueron positivos para aPS/PT IgG. El anticuerpo antifosfolípido más frecuente fue AL. Un paciente con PAM y uno con EB fueron positivos para aCL IgM; uno con PAM para anti-B2GP1 IgG, uno con PAN para anti-B2GP1 IgM.
ConclusionesLos anticuerpos aPS/PT son infrecuentes en pacientes con PAN inactiva de larga evolución.
Anti-phosphatidylserine/prothrombin (aPS/PT) antibodies have been described in cutaneous Polyarteritis Nodosa (PAN) in association with specific manifestations.
ObjectivesTo determine aPS/PT antibodies in patients with PAN and its correlation with clinical manifestations.
MethodsCross-sectional comparative study including PAN patients and 20 controls (10 Microscopic Polyangiitis [MPA] and 10 Behçet's disease [BD]). Clinical and demographic variables, treatment, serological markers, prognosis, activity and damage indexes were evaluated. aPS/PT, anti-cardiolipin (aCL), anti-beta 2 glycoprotein 1 (anti-B2GP1) antibodies, and lupus anticoagulant (LA) were determined.
ResultsFourteen patients with PAN were included, 11 (79%) women, with disease duration of 207 months, and mostly inactive disease. Only one patient with PAN and one with BD were positive for aPS/PT IgG. LA was the most frequent antibody identified. One patient with MPA and one with BD were positive for aCL IgM; one with MPA for anti-B2GP1 IgG, and one with PAN for anti-B2GP1 IgM.
ConclusionsaPS/PT antibodies are not frequent in patients with longstanding inactive PAN.
La poliarteritis nodosa (PAN) es una vasculitis necrosante que afecta predominantemente vasos de mediano calibre, no existen pruebas serológicas específicas para su diagnóstico1.
Los anticuerpos antifosfatidilserina/protrombina (aPS/PT) se han descrito en diversas vasculitis. Pocos estudios han investigado la prevalencia y significado de estos anticuerpos en PAN y se han enfocado principalmente al fenotipo cutáneo y/o etapas tempranas de la enfermedad2–4.
El objetivo del presente estudio fue determinar anticuerpos aPS/PT en pacientes con PAN sistémica y cutánea comparado con otras vasculitis, determinar si existe correlación con manifestaciones clínicas, otros anticuerpos antifosfolípidos, actividad y marcadores de inflamación.
Material y métodosEstudio comparativo transversal que incluyó pacientes mexicanos con PAN y controles con otras formas de vasculitis sistémicas (poliangitis microscópica [PAM]) y enfermedad de Behçet [EB]). Los pacientes fueron reclutados en la consulta externa de Reumatología de junio 2018 a mayo 2019 en el Instituto Nacional de Ciencias Médicas y Nutrición Salvador Zubirán. El diagnóstico de PAN se estableció con los criterios de 1990 del American College ofRheumatology, algoritmo de la European Agency for the Evaluation of Medical Products (EMEA) y/o definición del Consenso de Chapel Hill 20125–7. Los pacientes con PAN de inicio en la infancia cumplían criterios de Ankara 2008 (EULAR/PRES/PRINTO)8. El diagnóstico de PAM se estableció con el algoritmo EMEA y/o definición del Consenso de Chapel Hill 20126,7, y el de EB con criterios del Grupo de Estudio Internacional para EB9. Se excluyeron pacientes con PAN asociada a hepatitis B, otra enfermedad autoinmune, cáncer, infección, o tratamiento con anticoagulantes.
Se recabaron variables clínicas y demográficas, estudios de angiografía y patología al diagnóstico. En los pacientes con PAN se evaluó pronóstico al diagnóstico mediante el FFS (Five-Factor Score)10; actividad de la enfermedad (Birmingham Vasculitis Activity Score [BVAS 2003])11 al diagnóstico y al reclutamiento; extensión de la afección (Disease Extent Index [DEI])12 y daño (Vasculitis Damage Index [VDI])13 al reclutamiento. Se recabaron variables serológicas (velocidad de sedimentación globular [VSG]) y proteína C reactiva [PCR]), así como tratamiento actual.
Se procesaron anticuerpos anticardiolipinas (aCL) (IgG e IgM; INOVA Diagnostics; San Diego, EUA) y anti-beta 2 glicoproteína 1 (anti-B2GP1) (IgG e IgM; Orgentec Diagnostika; Mainz, Alemania) mediante ELISA automatizado, considerando positivo valores mayores al percentil 99°. Se determinó anticoagulante lúpico (AL) en plasma mediante la prueba coagulométrica (LA1 screening/LA2 confirmatorio; Siemens), basados en ensayo de veneno de víbora de Russel diluido. Se determinaron anticuerpos aPS/PT (IgG e IgM; INOVADiagnostics; San Diego, EUA) usando un kit de ELISA comercial, con valores de referencia de acuerdo con el percentil 95° (IgG: ≤ 15,7 U/mL e IgM ≤ 19,5 U/mL)14. Las muestras fueron conservadas a -70°C y los anticuerpos se determinaron cuando finalizó el reclutamiento.
El Comité de Ética Institucional aprobó el protocolo y los participantes firmaron un consentimiento informado. Los procedimientos se realizaron de acuerdo con los estándares éticos y la Declaración de Helsinki de 1964.
Las variables continuas fueron expresadas como medianas con intervalo intercuartil (IQR); las categóricas con valores absolutos y porcentajes. Las diferencias entre pacientes con PAN y controles se evaluaron usando prueba t de Student o U de Mann-Whitney (variables continuas), y prueba de X2 o exacta de Fisher (variables categóricas). Los valores de p < 0,05 se consideraron significativos. Se utilizó Stata (Stata Corp; College Stations, Texas, EUA) versión 12.0 y GraphPad Prism software versión 8.0.
ResultadosSe incluyeron 14 pacientes con PAN con mediana de edad al diagnóstico de 28 años (IQR 15-42), 11 (79%) de género femenino; 10 (71%) pacientes tenían PAN sistémica, cuatro (29%) cutánea y cinco (36%) inicio en la infancia.
Al diagnóstico las manifestaciones clínicas más frecuentes fueron síntomas constitucionales y musculoesqueléticos en 12 (86%) pacientes cada uno; seguido de manifestaciones cutáneas en 10 (71%) y neuropatía periférica en seis (43%). Al diagnóstico, la mediana de BVAS fue de nueve puntos (IQR 5-14) y de FFS de 1 (IQR 0-1). La mitad de los pacientes a los que se les realizó angiografía presentaron microaneurismas y 10/11 (91%) tuvieron confirmación histopatológica de vasculitis. Ningún paciente tenía antecedentes de trombosis. Los pacientes con PAN presentaban una mediana de DEI de 6 (IQR 5-8) y de VDI de 1 (IQR 1-3).
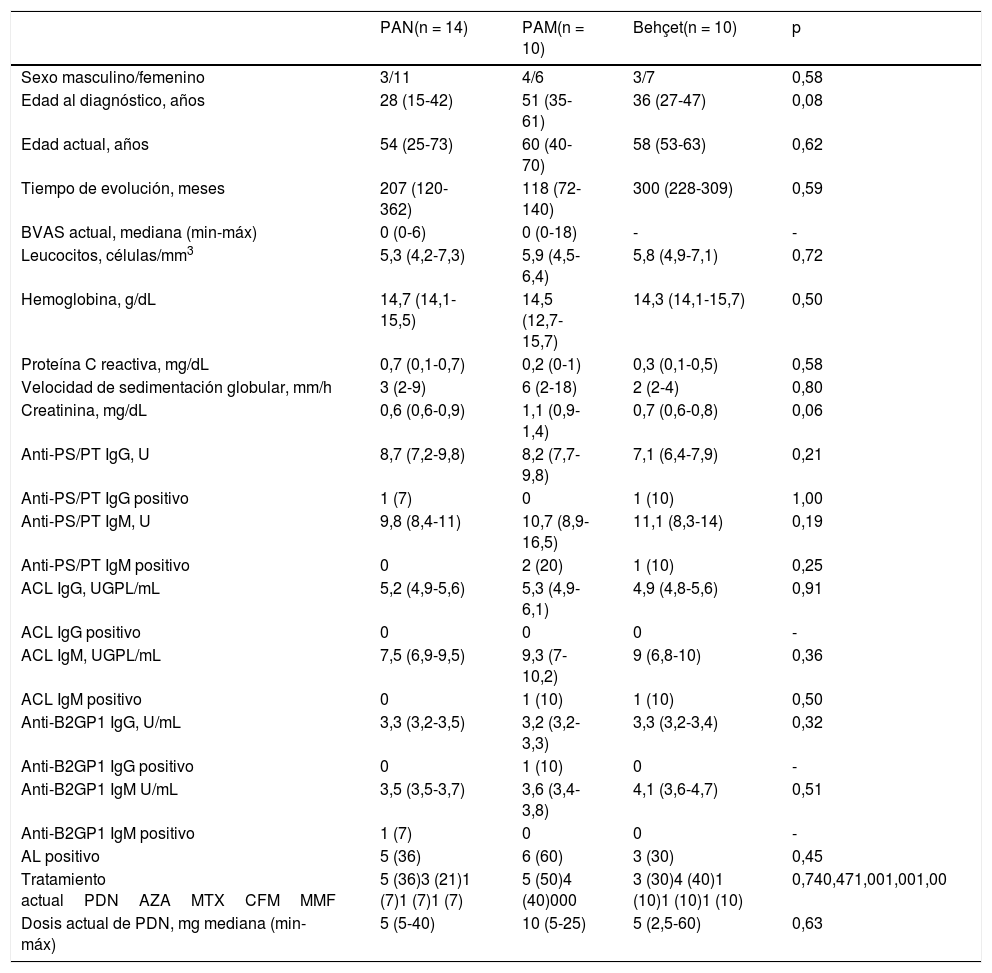
Se reclutaron 10 pacientes con PAM y 10 con EB como controles. La tabla 1 muestra las características demográficas y clínicas, así como anticuerpos antifosfolípidos en pacientes y controles. No hubo diferencias entre los grupos en cuanto a edad, sexo, duración de la enfermedad o tratamiento.
Análisis comparativo de los pacientes con PAN y grupo control con otras formas de vasculitis
| PAN(n = 14) | PAM(n = 10) | Behçet(n = 10) | p | |
|---|---|---|---|---|
| Sexo masculino/femenino | 3/11 | 4/6 | 3/7 | 0,58 |
| Edad al diagnóstico, años | 28 (15-42) | 51 (35-61) | 36 (27-47) | 0,08 |
| Edad actual, años | 54 (25-73) | 60 (40-70) | 58 (53-63) | 0,62 |
| Tiempo de evolución, meses | 207 (120-362) | 118 (72-140) | 300 (228-309) | 0,59 |
| BVAS actual, mediana (min-máx) | 0 (0-6) | 0 (0-18) | - | - |
| Leucocitos, células/mm3 | 5,3 (4,2-7,3) | 5,9 (4,5-6,4) | 5,8 (4,9-7,1) | 0,72 |
| Hemoglobina, g/dL | 14,7 (14,1-15,5) | 14,5 (12,7-15,7) | 14,3 (14,1-15,7) | 0,50 |
| Proteína C reactiva, mg/dL | 0,7 (0,1-0,7) | 0,2 (0-1) | 0,3 (0,1-0,5) | 0,58 |
| Velocidad de sedimentación globular, mm/h | 3 (2-9) | 6 (2-18) | 2 (2-4) | 0,80 |
| Creatinina, mg/dL | 0,6 (0,6-0,9) | 1,1 (0,9-1,4) | 0,7 (0,6-0,8) | 0,06 |
| Anti-PS/PT IgG, U | 8,7 (7,2-9,8) | 8,2 (7,7-9,8) | 7,1 (6,4-7,9) | 0,21 |
| Anti-PS/PT IgG positivo | 1 (7) | 0 | 1 (10) | 1,00 |
| Anti-PS/PT IgM, U | 9,8 (8,4-11) | 10,7 (8,9-16,5) | 11,1 (8,3-14) | 0,19 |
| Anti-PS/PT IgM positivo | 0 | 2 (20) | 1 (10) | 0,25 |
| ACL IgG, UGPL/mL | 5,2 (4,9-5,6) | 5,3 (4,9-6,1) | 4,9 (4,8-5,6) | 0,91 |
| ACL IgG positivo | 0 | 0 | 0 | - |
| ACL IgM, UGPL/mL | 7,5 (6,9-9,5) | 9,3 (7-10,2) | 9 (6,8-10) | 0,36 |
| ACL IgM positivo | 0 | 1 (10) | 1 (10) | 0,50 |
| Anti-B2GP1 IgG, U/mL | 3,3 (3,2-3,5) | 3,2 (3,2-3,3) | 3,3 (3,2-3,4) | 0,32 |
| Anti-B2GP1 IgG positivo | 0 | 1 (10) | 0 | - |
| Anti-B2GP1 IgM U/mL | 3,5 (3,5-3,7) | 3,6 (3,4-3,8) | 4,1 (3,6-4,7) | 0,51 |
| Anti-B2GP1 IgM positivo | 1 (7) | 0 | 0 | - |
| AL positivo | 5 (36) | 6 (60) | 3 (30) | 0,45 |
| Tratamiento actualPDNAZAMTXCFMMMF | 5 (36)3 (21)1 (7)1 (7)1 (7) | 5 (50)4 (40)000 | 3 (30)4 (40)1 (10)1 (10)1 (10) | 0,740,471,001,001,00 |
| Dosis actual de PDN, mg mediana (min-máx) | 5 (5-40) | 10 (5-25) | 5 (2,5-60) | 0,63 |
Los valores se representan como n (%) o mediana (p25-p75) si no se especifica lo contrario.
aCL: anticuerpos anticardiolipina; Anti-B2GP1: anticuerpos anti-beta 2 glicoproteína 1; aPS/PT: anticuerpos antifosfatidilserina/protrombina; BVAS: Birmingham Vasculitis Activity Score; AL: anticoagulante lúpico; PAM: poliangeítis microscópica; PAN: poliarteritis nodosa; PDN: prednisona; AZA: azatioprina; MTX: metotrexate; CFM: ciclofosfamida; MMF: mofetil micofenolato.
Al reclutamiento, los pacientes con PAN tenían duración de la enfermedad de 207 meses (IQR 120-362) y la mayoría presentaba enfermedad inactiva (mediana de BVAS 0 puntos [IQR 0-6]), niveles bajos de reactantes de fase aguda (PCR 0,7 mg/dL [IQR 0,1-0,7] y VSG 3 mm/hr [IQR 2-9]), y mediana de dosis de prednisona de 5 mg/día.
Algunos pacientes con PAN presentaban síntomas persistentes: constitucionales en dos (14%), musculoesqueléticos en cuatro (29%) y neuropatía periférica en cuatro (29%).
Un paciente con PAN y uno con EB fueron positivos para aPS/PT IgG, y ningún paciente con PAN tuvo aPS/PT IgM positivo; sin embargo, este anticuerpo estuvo presente en dos pacientes con PAM y uno con EB. El AL fue el anticuerpo más frecuente, en cinco (36%) pacientes con PAN, seis (60%) con PAM y tres (30%) con EB (p = 0,45). Un paciente con PAM y uno con EB tuvieron aCL IgM positivos; uno con PAM anti-B2GP1 IgG positivo y uno con PAN anti-B2GP1 IgM positivo. La figura 1 muestra los títulos de anticuerpos aPS/PT IgG e IgM en pacientes con PAN y controles.
 y aPS/PT IgM (b) en poliarteritis nodosa (PAN) y controles con poliangeítis microscópica (PAM) y enfermedad de Behçet (EB).")
La única paciente con PAN y aPS/PT IgG positivos también tenía positividad para AL. Al diagnóstico de PAN presentó mononeuropatía múltiple, fiebre, síntomas musculoesqueléticos, nódulos subcutáneos y livedo reticularis. Al momento del reclutamiento, 11 meses posterior al diagnóstico, presentaba síntomas constitucionales y musculoesqueléticos, además de neuropatía (BVAS 6 puntos).
DiscusiónEl presente estudio analizó la presencia de anticuerpos aPS/PT en pacientes con PAN predominantemente de fenotipo sistémico y su asociación con manifestaciones clínicas. Ningún paciente con PAN fue positivo para aPS/PT IgM y sólo uno presentó aPS/PT IgG positivo. Estos resultados son contrarios a estudios previos en los cuales la presencia de anticuerpos aPS/PT y otros anticuerpos antifosfolípidos se han descrito en pacientes con PAN2–4.
Kawakami et al. encontraron que 13 de 16 (81.3%) pacientes con PAN cutánea tenían aPS/PT IgM positivos, con títulos más altos que controles con lupus eritematoso generalizado. En este estudio, pacientes con PAM y controles sanos no presentaron positividad para dichos anticuerpos; además, se reportó una correlación positiva entre anticuerpos aPS/PT IgM y niveles de PCR en pacientes que también tenían AL positivo2.
El mismo grupo también describió la correlación entre manifestaciones cutáneas (livedo racemosa y placas inflamatorias) y la presencia de AL y anticuerpos aCL y aPS/PT en pacientes con PAN cutánea. Encontraron niveles más altos de anticuerpos aPS/PT IgM en pacientes con livedo racemosa y de aPS/PT IgG y aCL IgG en aquellos con placas inflamatorias3.
Por otro lado, Okano et al., estudiaron pacientes con PAN con manifestaciones cutáneas activas y determinaron anticuerpos aPS/PT y citocinas antes y después del tratamiento con ciclofosfamida y glucocorticoides. En dicho estudio se encontró que los anticuerpos aPS/PT y los niveles de IL-2 después del tratamiento fueron menores comparado con los niveles previos al tratamiento4.
Lo anterior indica que la presencia de anticuerpos aPS/PT en pacientes con PAN pudiera estar condicionada por la duración de la enfermedad, fenotipo clínico y presencia de actividad, ya que la mayoría de los pacientes con reactividad para dichos autoanticuerpos presentaban PAN cutánea, activa, de reciente inicio, virgen a tratamiento2,3.
Los pacientes incluidos en el presente estudio tenían evolución de la enfermedad prolongada, eran predominantemente del fenotipo sistémico, y mostraban enfermedad inactiva al momento del reclutamiento; además, manifestaciones cutáneas como livedo reticularis o racemosa no estaban presentes, lo que pudiera explicar la ausencia de aPS/PT en nuestra cohorte.
El presente trabajo exhibe limitantes. El tamaño de muestra es pequeño y no se consideraron controles sanos para comparar los títulos de anticuerpos. Los pacientes tenían una duración de la enfermedad prolongada, aunado a que la mayoría se encontraba en remisión y había recibido tratamiento previamente; y finalmente, el hecho de que los anticuerpos se determinaron en una sola ocasión. Consideramos, sin embargo, que el presente estudio refleja un panorama de pacientes con PAN ambulatorios, incluye pacientes con fenotipo predominantemente sistémico y con esto agrega información a reportes previos de anticuerpos aPS/PT en PAN cutánea. Así mismo, se incluyeron pacientes con otras formas de vasculitis como controles y se determinó un perfil completo de anticuerpos antifosfolípidos.
En conclusión, los resultados del presente estudio sugieren que los anticuerpos aPS/PT son infrecuentes en pacientes con PAN de larga evolución en remisión. Para confirmar estos resultados, son necesarios estudios longitudinales, con mayor número de pacientes con fenotipos cutáneo y sistémico, enfermedad activa y en remisión, previo y posterior a tratamiento.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.
FinanciaciónEste trabajo no ha recibido ningún tipo de financiación.
Los autores agradecen a Isela Chan Campos, Darinel Hernández Hernández y Andrés Valencia Martínez su valioso apoyo en la toma de muestras y en la determinación de anticoagulante lúpico.


