





Mejorar el nivel de conocimiento sobre los medicamentos biosimilares y generar un marco consensuado sobre su uso.
MétodosEstudio cualitativo. Se seleccionó un grupo multidisciplinar de expertos en medicamentos biosimilares (una dermatóloga, un farmacéutico de hospital, un reumatólogo y un gastroenterólogo) que definieron los apartados y los temas del documento. Se realizó una revisión narrativa de la literatura en Medline para identificar artículos sobre los medicamentos biosimilares. Se seleccionaron revisiones sistemáticas de la literatura, estudios controlados pre-clínicos, clínicos y en vida real. Con esta información se generaron varios principios generales y recomendaciones. El grado de acuerdo con los mismos se estableció mediante un Delphi que se extendió a 66 profesionales de la salud que votaron de 1 (totalmente en desacuerdo) a 10 (totalmente de acuerdo). Se definió acuerdo si al menos el 70% de los participantes votaron ≥7.
ResultadosLa revisión de la literatura incluyó 555 artículos. Se votaron un total de 10 principios generales y recomendaciones. Todos alcanzaron el nivel de acuerdo establecido en el Delphi. El documento incluye datos sobre las características principales de los medicamentos biosimilares (definición, desarrollo, aprobación, extrapolación de indicaciones, intercambiabilidad, financiación y trazabilidad); sobre la evidencia publicada (biosimilitud, eficacia, efectividad, seguridad, inmunogenicidad, eficiencia, switch); sobre barreras y facilitadores a su uso, y datos sobre la información para pacientes.
ConclusionesLos medicamentos biosimilares autorizados reúnen todas las características de calidad, eficacia y seguridad. Además, ayudan significativamente a mejorar el acceso de los pacientes a las terapias biológicas y contribuyen a la sostenibilidad de los sistemas sanitarios.
To improve knowledge about biosimilar medicines and to generate a consensus framework on their use.
MethodsQualitative study. A multidisciplinary group of experts in biosimilar medicines was established (1dermatologist, 1hospital pharmacist, 1rheumatologist, and 1gastroenterologist) who defined the sections and topics of the document. A narrative literature review was performed in Medline to identify articles on biosimilar medicines. Systematic reviews, controlled, pre-clinical, clinical, and real-life studies were selected. Based on the results of the review, several general principles and recommendations were generated. The level of agreement was tested in a Delphi that was extended to 66 health professionals who voted from 1 (totally disagree) to 10 (totally agree). Agreement was defined if at least 70% of the participants voted ≥7.
ResultsThe literature review included 555 articles. A total of 10 general principles and recommendations were voted upon. All reached the level of agreement established. The document includes data on the main characteristics of biosimilar medicines (definition, development, approval, indication extrapolation, interchangeability, financing, and traceability); published evidence (biosimilarity, efficacy, effectiveness, safety, immunogenicity, efficiency, switch); barriers and facilitators to its use; and data on information for patients.
ConclusionsAuthorized biosimilar medicines meet all the characteristics of quality, efficacy, and safety. They also significantly help improve patient access to biological therapies and contribute to health system sustainability.
En nuestro país, los medicamentos están sujetos a una estricta normativa y control para garantizar su seguridad y su eficacia.
El proceso por el que se aprueba un medicamento tradicional, de síntesis química, se ha ido perfeccionando a lo largo de las décadas y actualmente deja poco espacio para la improvisación. Sin embargo, la llegada de los medicamentos biológicos ha supuesto un gran cambio, ya que ni los fabricantes ni las agencias reguladoras habían tratado con moléculas estructuralmente tan grandes y complejas. Además, y a diferencia de las versiones genéricas de los medicamentos tradicionales, la posibilidad de copiar la molécula de un biológico, una vez que su patente ha expirado, se ve dificultada por la necesidad de desarrollar un proceso de fabricación alternativo, ya que el procedimiento original está protegido por patentes adicionales que tienen una validez sustancialmente mayor1.
El gran tamaño molecular de los medicamentos biológicos, combinado con la variabilidad asociada a las fuentes biológicas, determina que el paradigma de la fabricación de los fármacos convencionales no sea aplicable. Debido a esta variabilidad natural de la fuente biológica y al proceso de fabricación específico de cada fabricante, pueden aparecer ligeras diferencias en la composición entre el medicamento biosimilar y su medicamento de referencia, de igual manera que aparecen diferencias entre lotes del mismo medicamento biológico de referencia2.
Actualmente en España disponemos de más de 50 medicamentos biosimilares correspondientes a 16 principios activos, 4 de ellos para enfermedades inmunomediadas reumatológicas, dermatológicas y del aparato digestivo: adalimumab (ADA), etanercept (ETN), infliximab (IFX) y rituximab (RTX). El primer medicamento biosimilar para este grupo de enfermedades se aprobó en 2015 y fue de IFX. Desde entonces los medicamentos biosimilares son ampliamente utilizados en nuestro país3.
Sin embargo, por otro lado, datos provenientes de una encuesta nacional sugieren que el nivel de conocimiento sobre múltiples aspectos (algunos muy relevantes) de los medicamentos biosimilares, como su desarrollo, los fundamentos, el acceso o el uso en la práctica clínica, es muy bajo4. Además, este trabajo mostró la gran variabilidad existente con su uso en los hospitales españoles4. Es por ello que los objetivos de este trabajo fueron el de mejorar el conocimiento y el uso de los medicamentos biosimilares en enfermedades inmunomediadas, y el de generar un marco consensuado sobre el uso de biosimilares. Para ello hemos realizado una extensa revisión de la literatura y hemos contado con la opinión de un grupo multidisciplinar de expertos. Este documento pretende ser una referencia para profesionales de la salud implicados en el manejo de pacientes con enfermedades inmunomediadas con medicamentos biosimilares.
MetodologíaDiseño del estudioEstudio cualitativo. Se siguió la metodología de grupos nominales y Delphi, con ayuda de una revisión narrativa de la literatura. El proyecto se efectuó en plena conformidad con los principios establecidos en la Declaración de Helsinki, referente a la investigación médica en seres humanos, en su última versión, y de acuerdo con la normativa aplicable sobre buena práctica clínica.
Selección de participantes y primera reunión de grupo nominalEn primer lugar se seleccionó un grupo multidisciplinar formado por cuatro profesionales de la salud con gran experiencia y conocimiento en medicamentos biosimilares (una dermatóloga, un farmacéutico de hospital, un reumatólogo y un gastroenterólogo). Tras ello, y con ayuda metodológica, se definieron los objetivos, el alcance, los usuarios y los apartados a desarrollar del documento. Estos incluyen:
- 1.
Características principales de los medicamentos biosimilares (definición, desarrollo, aprobación, extrapolación de indicaciones, intercambiabilidad, financiación y trazabilidad).
- 2.
La evidencia (biosimilitud, eficacia, efectividad, seguridad, inmunogenicidad, eficiencia, switch).
- 3.
Barreras y facilitadores a su uso.
- 4.
Información para pacientes. En base a esto se realizó la revisión de la literatura.
Con la ayuda de una experta documentalista se realizó una revisión narrativa de la literatura. Se interrogó Medline utilizando la herramienta Clinical Queries de Pubmed y búsquedas individuales con lenguaje controlado (Mesh) y con términos de texto libre (hasta julio de 2021). Nuestro objetivo fue identificar artículos que analizasen las características principales y el uso de medicamentos biosimilares en enfermedades inmunomediadas de las especialidades de reumatología, dermatología y gastroenterología. Se seleccionaron revisiones sistemáticas de la literatura, así como estudios de desarrollo pre-clínico, ensayos clínicos aleatorizados (ECA) y estudios de vida real. Dos revisores seleccionaron de forma independiente los artículos (primero por título y resumen, y después tras leer los artículos completos en detalle) y recopilaron datos. Se generaron tablas de evidencia y resultados. La calidad de los estudios se evaluó mediante la escala de Oxford de 20115. Con esta información el coordinador generó una serie de principios generales y recomendaciones preliminares.
Segunda reunión de grupo nominalEn la segunda reunión de grupo nominal se presentaron y discutieron los resultados de la revisión narrativa de la literatura, así como los principios generales y recomendaciones provisionales. Con ello se definieron las recomendaciones definitivas que fueron sometidas a un proceso Delphi.
DelphiLas recomendaciones se votaron en un Delphi para establecer el grado de acuerdo con las mismas. Esta se realizó on-line, fue anónima, y se envió a 66 profesionales de la salud (médicos especialistas y farmacéuticos de hospital). El grado de acuerdo se expresaba mediante votación en una escala Likert de 1 (totalmente en desacuerdo) a 10 (totalmente de acuerdo). Se definió acuerdo si al menos el 70% de los participantes votaban ≥7. Las recomendaciones con un grado de acuerdo inferior al 70% fueron evaluadas y, si procedía, re-editadas y votadas en una segunda ronda Delphi. En la primera ronda Delphi se permitió incluir nuevas recomendaciones.
Edición del documento finalCon la revisión de la literatura, las decisiones del grupo nominal y el Delphi se redactó el documento definitivo. El documento final se distribuyó entre los expertos para su valoración final y los últimos comentarios.
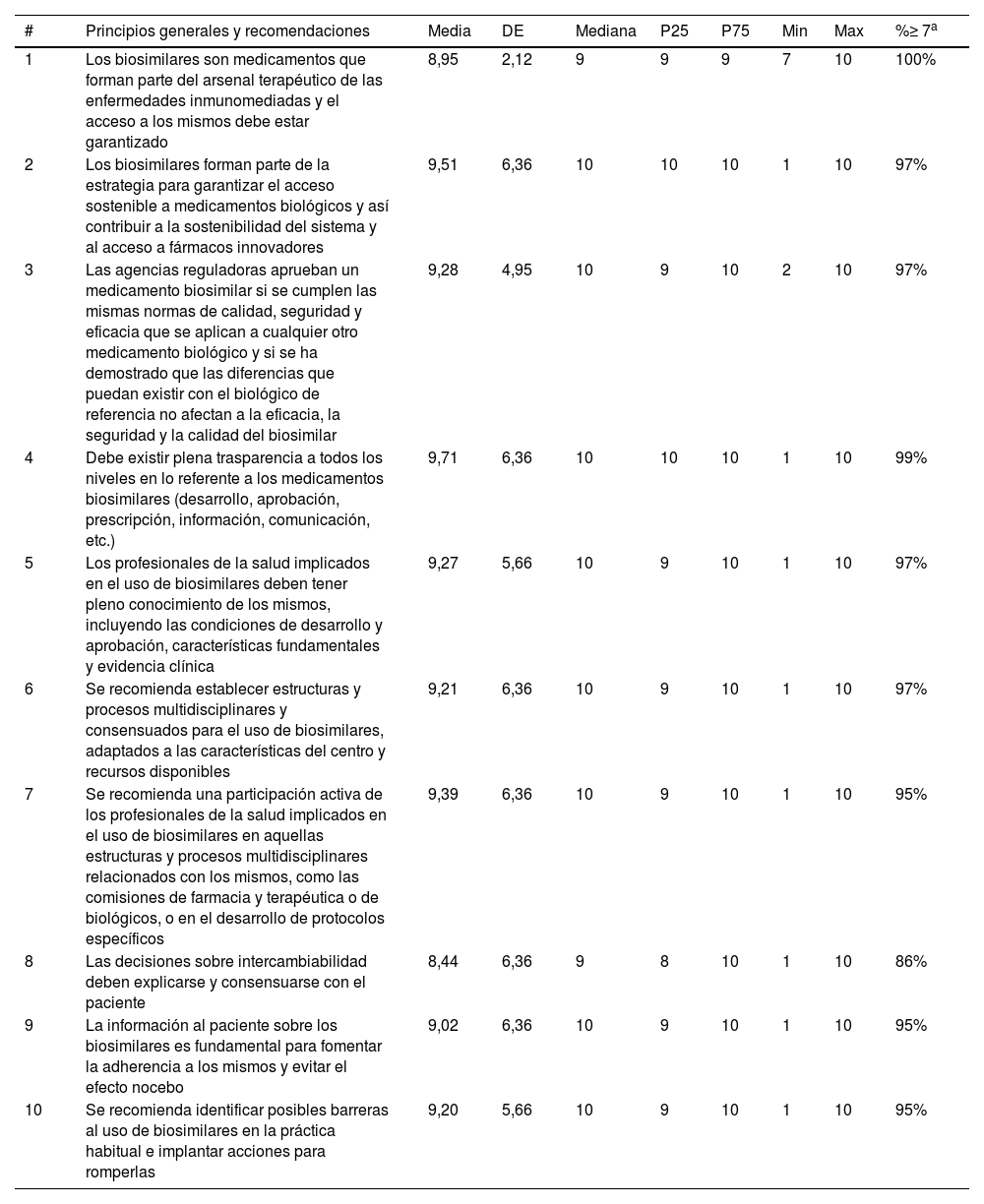
ResultadosRevisión narrativa de la literatura y DelphiLa revisión encontró más de 500 artículos. Con esta información y la opinión de los expertos se generaron un total de 10 principios generales y recomendaciones que alcanzaron un altísimo nivel de acuerdo (tabla 1).
Resultados del Delphi
| # | Principios generales y recomendaciones | Media | DE | Mediana | P25 | P75 | Min | Max | %≥ 7a |
|---|---|---|---|---|---|---|---|---|---|
| 1 | Los biosimilares son medicamentos que forman parte del arsenal terapéutico de las enfermedades inmunomediadas y el acceso a los mismos debe estar garantizado | 8,95 | 2,12 | 9 | 9 | 9 | 7 | 10 | 100% |
| 2 | Los biosimilares forman parte de la estrategia para garantizar el acceso sostenible a medicamentos biológicos y así contribuir a la sostenibilidad del sistema y al acceso a fármacos innovadores | 9,51 | 6,36 | 10 | 10 | 10 | 1 | 10 | 97% |
| 3 | Las agencias reguladoras aprueban un medicamento biosimilar si se cumplen las mismas normas de calidad, seguridad y eficacia que se aplican a cualquier otro medicamento biológico y si se ha demostrado que las diferencias que puedan existir con el biológico de referencia no afectan a la eficacia, la seguridad y la calidad del biosimilar | 9,28 | 4,95 | 10 | 9 | 10 | 2 | 10 | 97% |
| 4 | Debe existir plena trasparencia a todos los niveles en lo referente a los medicamentos biosimilares (desarrollo, aprobación, prescripción, información, comunicación, etc.) | 9,71 | 6,36 | 10 | 10 | 10 | 1 | 10 | 99% |
| 5 | Los profesionales de la salud implicados en el uso de biosimilares deben tener pleno conocimiento de los mismos, incluyendo las condiciones de desarrollo y aprobación, características fundamentales y evidencia clínica | 9,27 | 5,66 | 10 | 9 | 10 | 1 | 10 | 97% |
| 6 | Se recomienda establecer estructuras y procesos multidisciplinares y consensuados para el uso de biosimilares, adaptados a las características del centro y recursos disponibles | 9,21 | 6,36 | 10 | 9 | 10 | 1 | 10 | 97% |
| 7 | Se recomienda una participación activa de los profesionales de la salud implicados en el uso de biosimilares en aquellas estructuras y procesos multidisciplinares relacionados con los mismos, como las comisiones de farmacia y terapéutica o de biológicos, o en el desarrollo de protocolos específicos | 9,39 | 6,36 | 10 | 9 | 10 | 1 | 10 | 95% |
| 8 | Las decisiones sobre intercambiabilidad deben explicarse y consensuarse con el paciente | 8,44 | 6,36 | 9 | 8 | 10 | 1 | 10 | 86% |
| 9 | La información al paciente sobre los biosimilares es fundamental para fomentar la adherencia a los mismos y evitar el efecto nocebo | 9,02 | 6,36 | 10 | 9 | 10 | 1 | 10 | 95% |
| 10 | Se recomienda identificar posibles barreras al uso de biosimilares en la práctica habitual e implantar acciones para romperlas | 9,20 | 5,66 | 10 | 9 | 10 | 1 | 10 | 95% |
DE: desviación estándar; Max: máximo; Min: mínimo; P25: percentil 25; P75: percentil 75.
Los expertos consideran que los medicamentos biosimilares forman parte de la estrategia para garantizar el acceso sostenible a medicamentos biológicos y a otros medicamentos innovadores, contribuyendo así a la sostenibilidad del sistema. Por ello su acceso debe estar garantizado.
Además, se insiste en que las agencias reguladoras aprueban un medicamento biosimilar solo si se cumplen las mismas normas de calidad, seguridad y eficacia que se aplican a otros medicamentos biológicos y una vez que se ha demostrado que las diferencias que puedan existir con el biológico de referencia no afectan a estos parámetros.
Por ello, es importante que los profesionales de la salud implicados en el uso de medicamentos biosimilares tengan pleno conocimiento de sus características y participen en todas las estructuras y los procesos relacionados con los mismos, como las comisiones de farmacia y terapéutica o de biológicos, o en el desarrollo de protocolos específicos.
Igualmente los expertos destacan el papel del paciente con el uso de los medicamentos biosimilares. Así, por ejemplo, hay gran acuerdo en que las decisiones sobre intercambiabilidad deben explicarse y consensuarse con el paciente, así como en la importancia de la información que hay que ofrecerles.
A continuación se explican pormenorizadamente cada uno de los apartados evaluados en la revisión y Delphi.
Conceptos, definiciones y desarrollo de los medicamentos biosimilares• ¿Qué es un medicamento biológico?Los medicamentos biológicos son aquellos que contienen uno o más principios activos producidos o derivados de una fuente biológica de origen recombinante o extractivo6. Su composición química es muy variada, pudiendo incluir proteínas, hidratos de carbono, ácidos nucleicos, o combinaciones de estas sustancias, o incluso estar formados por seres vivos completos, como células o tejidos6. Los medicamentos biológicos pueden obtenerse de múltiples fuentes naturales: humanos, animales o microorganismos7.
• ¿Qué es un medicamento biosimilar?Las principales características de los medicamentos biosimilares se recogen en la tabla 2.
Características principales de los medicamentos biosimilares
| # | Característica | Comentarios |
|---|---|---|
| 1 | Son muy similares al medicamento de referencia | • El biosimilar tiene propiedades físicas, químicas y biológicas muy similares al medicamento de referencia |
| 2 | La variabilidad del medicamento biosimilar se mantiene dentro de unos límites estrictos | • Todos los medicamentos biológicos (biosimilares y de referencia) por su naturaleza inherente presentan cierto grado de variabilidad (diferencias en su composición)• Solo se permite un pequeño margen de variabilidad• Esto se consigue mediante un proceso de fabricación consistente, que garantice que todos los lotes del medicamento tienen calidad probada |
| 3 | No presentan ninguna diferencia clínicamente significativa en relación con el medicamento de referencia | • Los estudios clínicos en los que se basa la aprobación de un biosimilar confirman que las diferencias no tendrán efectos sobre la seguridad ni sobre la eficacia |
| 4 | La EMA sigue normas de calidad, seguridad y eficacia muy estrictas para la aprobación de medicamentos biosimilares | • Para que se aprueben los biosimilares se tienen que cumplir las mismas normas de calidad, seguridad y eficacia que se aplican a cualquier otro medicamento |
EMA: European Medicines Agency.
Según la European Medicines Agency (EMA), un medicamento biosimilar es un medicamento biológico muy similar a otro medicamento biológico ya comercializado en la Unión Europea (UE), denominado «medicamento de referencia», y frente al cual demuestra biosimilitud7,8. La biosimilitud es la propiedad de un medicamento para mostrar similitud y falta de diferencias significativas en términos de calidad, eficacia y seguridad respecto a un medicamento de referencia con el que se ha comparado.
Aunque la estructura química básica del medicamento biosimilar pueda ser idéntica a la del de referencia, una vez que una proteína se «traduce», experimenta modificaciones adicionales (glucosilación, sulfatación, metilación, etc.). Estas modificaciones serán únicas para cada molécula en particular, por lo que no habrá dos totalmente idénticas en un vial de cualquier medicamento biosimilar, y de igual manera aparecen diferencias entre lotes del mismo medicamento biológico de referencia. Por ello, cada medicamento biológico presenta una determinada disposición de atributos críticos de calidad. Estos atributos son propiedades fisicoquímicas y biológicas, algunas de las cuales son más sensibles que otras a las variaciones, e incluyen el tamaño, la carga molecular y la glucosilación. Todos ellos pueden modificarse mediante cambios en el proceso, por ejemplo, en el tipo de célula utilizada en el proceso de producción o por las condiciones de cultivo, la temperatura del pH, etc.
Es por ello que, durante el proceso de fabricación del medicamento biosimilar, siempre se realizan controles rigurosos para garantizar que las pequeñas diferencias existentes no afectan al funcionamiento del medicamento ni a su seguridad. Es decir, se garantiza que estas diferencias no son clínicamente significativas desde el punto de vista de la fisicoquímica, de la eficacia o de la seguridad9.
• ¿Cómo se aprueban los medicamentos biosimilares?Todos los medicamentos producidos mediante biotecnología deben ser autorizados en la UE a través de la EMA mediante el denominado «procedimiento centralizado». Es decir, un único expediente de registro es presentado a la EMA y es evaluado por los comités científicos de medicamentos de uso humano y de seguridad de la EMA (CHMP)10.
Cuando se solicita autorización a la EMA de un biosimilar, el CHMP, así como los expertos en medicamentos biológicos de la UE (grupo de trabajo de fármacos biológicos) y especialistas en biosimilares (grupo de trabajo de biosimilares), evalúan los estudios aportados para analizar si demuestra biosimilitud.
Esta demostración de biosimilitud se realiza a través de una detallada comparación (tabla 3). Esta es un ejercicio de comparabilidad directa entre el medicamento biosimilar y su medicamento de referencia, que comprueba que las pequeñas diferencias en la estructura y la función (fisicoquímicas o de actividad biológica) que puedan existir entre ambos no afectan a la eficacia, a la seguridad y a la calidad del medicamento biosimilar6. Esta comparabilidad no solo se realiza con los medicamentos biosimilares, también con los medicamentos de referencia cuando estos experimentan modificaciones en sus procesos de producción o desarrollan nuevas formulaciones galénicas. Esta evaluación comparativa se hace sobre una o varias indicaciones sensibles, es decir, sobre la población donde se pueden detectar de mejor manera las diferencias en el rendimiento clínico relacionadas con el medicamento biosimilar.
Proceso de comparabilidad de un medicamento biosimilar y su medicamento de referencia
| # | Etapa | Elementos evaluados |
|---|---|---|
| 1 | Comparativa fisicoquímica | • Ensayos fisicoquímicos mediante técnicas analíticas para garantizar la biosimilitud estructural entre ambos medicamentos• Se analizan y comparan los siguientes atributos: estructura primaria, estructuras de orden superior, contenido, pureza (agregados y fragmentos), isoformas (variantes de carga), glucosilación, etc. |
| 2 | Comparativa preclínica | • Estudios in vitro, ex vivo y, si se considera oportuno, in vivo para garantizar la misma actividad biológica / farmacológica• Se analizan y comparan las funciones biológicas más relevantes para su acción terapéutica y su toxicidad: unión a receptores o a sus dianas biológicas, transducción de señales biológicas, viabilidad celular, etc. |
| 3 | Comparativa clínica | • Estudios de bioequivalencia, modelos farmacodinámicos validados, ensayos clínicos, etc., para garantizar el mismo comportamiento farmacocinético, farmacodinámico y clínico• Se analizan y se comparan la eficacia, la seguridad, la inmunogenicidad, etc. |
Cuando el CHMP emite una opinión favorable sobre un medicamento biosimilar, la UE se encarga de aprobar la comercialización del medicamento, quedando este autorizado automáticamente en todo el territorio de la UE. En la página web de la EMA se publican los informes EPAR (European Public Assessment Report) con el resumen de la evidencia científica que fundamenta la autorización del medicamento biosimilar.
Además, debido a la complejidad y a la heterogeneidad de los medicamentos biosimilares, la UE ha desarrollado un marco regulatorio específico basado en unos principios diferentes y más complejos que para los medicamentos genéricos. Las directivas y las guías aplicables a los medicamentos biosimilares también están disponibles en la página web de la EMA y se resumen en la tabla 4.
Documentación de carácter regulatorio de la EMA sobre biosimilares
| Tema | Documento | Última actualización |
|---|---|---|
| Principios generales | Guideline on similar biological medicine products | CHMP/437/04 Rev 1 octubre de 2014 / abril de 2015 |
| Calidad | Guideline on similar biological medicinal product containing biotechnology-derived proteins as active substance: quality issues | CHMP/BWP/247713/2012 mayo de 2014 / diciembre de 2014 |
| Estudios clínicos y no clínicos | Guideline on similar biological medicinal products containing biotechnology-derived proteins as active substance: non-clinical and clinical issue | CHMP/BMWP7/42832/2005 Rev 1 diciembre de 2014 / julio de 2015 |
EMA: European Medicines Agency.
En España, además de la aprobación de la EMA, previo a su comercialización debe ser emitida una resolución favorable de financiación con cargo al Sistema Nacional de Salud por parte del Ministerio de Sanidad, Consumo y Bienestar Social y, en su caso, ser fijado el precio por parte de la Comisión Interministerial de Precios.
• ¿Qué es la extrapolación de indicaciones?La extrapolación de indicaciones es la extensión de los datos relativos a la eficacia y a la seguridad de una indicación terapéutica para la cual el medicamento biosimilar ha sido clínicamente probado a otra indicación terapéutica autorizada para el medicamento de referencia.
Tras superar satisfactoriamente los estudios en la indicación o indicaciones más sensibles, se extrapolan al biosimilar aquellas otras indicaciones del medicamento de referencia que los evaluadores consideran oportuno a la vista de los resultados del ejercicio de comparabilidad. La extrapolación de datos a otras indicaciones siempre está basada en los datos científicos obtenidos en sólidos estudios de comparabilidad7.
La extrapolación es un principio científico consolidado que se ha utilizado durante muchos años en medicina, incluyendo a los medicamentos biológicos de referencia11. La extrapolación se realiza, por ejemplo, cuando un medicamento biológico con varias indicaciones autorizadas experimenta cambios importantes en su proceso de fabricación (nuevo lugar de fabricación, desarrollo de nuevas formulaciones galénicas, etc.). El efecto potencial de estos cambios sobre el rendimiento clínico del medicamento biológico es exhaustivamente evaluado mediante estudios de comparabilidad (estudios in vitro y de calidad). En caso de necesitarse estudios clínicos, estos se realizan en una indicación relevante y, sobre la base de todos estos datos, suele ser posible la extrapolación a las otras indicaciones.
• ¿Qué es la intercambiabilidad?La intercambiabilidad hace referencia a la posibilidad de intercambiar un medicamento por otro que se espera que tenga el mismo efecto clínico. Esto podría significar cambiar un medicamento de referencia por un medicamento biosimilar (o viceversa), o reemplazar un medicamento biosimilar por otro.
Este intercambio puede realizarse mediante un switch, en el que el cambio se realiza por decisión del prescriptor, o la sustitución, en el que el cambio se realiza de manera automática a nivel farmacéutico sin consultar con el prescriptor.
La EMA no incluye recomendaciones sobre la intercambiabilidad con el medicamento de referencia. Aunque aconsejan involucrar a los prescriptores en la decisión final, la postura conjunta de la EMA y la Comisión Europea es que sean los Estados miembros los que decidan si los medicamentos biológicos y sus respectivos biosimilares pueden ser intercambiables12.
En España, la orden SCO/2874/2007 permite el switch pero impide la sustitución automática cuando no hay un consenso previo con el prescriptor13, en línea con el posicionamiento de las principales sociedades científicas de reumatología, dermatología y oncología del país9,14,15. Sin embargo, esta orden hace referencia al artículo 86 de la Ley 29/2006, de 26 de julio, de garantías y uso racional de los medicamentos y productos sanitarios, artículo que está dentro del capítuloIV, que se refiere al uso racional de medicamentos en las oficinas de farmacia. Por tanto, no aplica al ámbito hospitalario, donde cada Comisión de Farmacia y Terapéutica (CFT) establece si se puede intercambiar un biosimilar y los criterios a aplicar mediante el consenso de todas las partes interesadas (médicos, farmacéuticos hospitalarios, farmacéuticos de atención primaria, gestores sanitarios y pacientes). De este modo, se permite el intercambio en el hospital si ha sido aprobado por las CFT de los hospitales, los comités autonómicos y el médico, que tiene representación en estos comités16.
• ¿Cómo se financian los medicamentos biosimilares?En general, la decisión de financiación pública de los medicamentos va de la mano de una evaluación de estos. En este sentido, los medicamentos biosimilares no son una excepción y siguen las mismas pautas de evaluación económica que el resto de los medicamentos biológicos. Respecto a la financiación de los medicamentos, hay múltiples factores que pueden influir en la determinación de su precio por parte de las autoridades (efectividad clínica incremental, coste-efectividad e impacto presupuestario, carga de la enfermedad y necesidad médica no cubierta, tamaño de la población objetivo, precios de referencia internos, precios de referencia internacionales, coste de producción, carácter innovador del producto, consideraciones éticas y de equidad, contribución al PIB y grupos de presión). Estos factores también son en cierta medida aplicables a los medicamentos biosimilares, si bien su referenciación al precio de los medicamentos de referencia es un elemento principal.
• ¿Qué es la trazabilidad?Se define como la habilidad para trazar y seguir un medicamento a través de todas sus etapas de producción, distribución y utilización. La trazabilidad, como mecanismo para el seguimiento de un medicamento a lo largo de su «vida», ha sido ampliamente reconocida como un elemento fundamental en la seguridad del paciente.
Otro de los requisitos a destacar de los medicamentos biosimilares para garantizar una correcta monitorización de su seguridad es la trazabilidad tanto de las prescripciones como de las administraciones a los pacientes que se hace con los mismos.
Evidencia sobre los medicamentos biosimilaresEs importante señalar en primer lugar que mientras que en los ECA de los biológicos originales el objetivo es demostrar el beneficio clínico y la seguridad en los pacientes, el objetivo de los ECA de los medicamentos biosimilares es el de excluir diferencias clínicamente relevantes específicas del producto (estudios de no inferioridad)8,17-22. Pero además actualmente contamos con muchos datos publicados de vida real23-26.
A continuación resumimos los principales datos de la evidencia con el uso de los medicamentos biosimilares aprobados para las enfermedades inmunomediadas de origen reumatológico, dermatológico y del aparato digestivo. Más concretamente, sobre IFX, ADA, ETN y RTX biosimilares. En el material suplementario se muestra un resumen de los ECA, las indicaciones aprobadas, las variables de desenlace utilizadas y si se ha estudiado el switch.
• Eficacia, efectividad, inmunogenicidad y seguridadLa eficacia de los medicamentos biosimilares en enfermedades inmunomediadas ha sido ampliamente demostrada en ECA y recogida en distintas revisiones sistemáticas y metaanálisis17-21. En estos estudios se ha mostrado que los medicamentos biosimilares son muy parecidos a los de referencia en términos de estructura, propiedades fisicoquímicas y biológicas, farmacocinética, eficacia, seguridad e inmunogenicidad. En el caso, por ejemplo, de pacientes con artritis reumatoide, no se han encontrado diferencias estadísticamente significativas en el ACR20, el ACR50 o el ACR70 a las 12, 24 y 52 semanas entre los medicamentos biosimilares y los de referencia, así como en la tasa de acontecimientos adversos graves17-19. Tampoco en las espondiloartritis, incluyendo la artritis psoriásica20,21. En pacientes con psoriasis cutánea, por otro lado, también se ha demostrado su bioequivalencia, su eficacia y su seguridad22.
En relación con la enfermedad inflamatoria intestinal, múltiples estudios observacionales han demostrado la efectividad y la seguridad de IFX y ADA biosimilar23,24.
Igualmente, se han publicado datos de estudios observacionales en otras enfermedades inmunomediadas cuyos resultados están en línea con los obtenidos en los ECA25,26.
• EficienciaEl impacto en el gasto farmacéutico derivado del acceso de medicamentos biológicos al mercado es significativo y creciente27,28. Su incuestionable valor clínico viene acompañado de un precio habitualmente más elevado que el de fármacos de síntesis química. La introducción de los medicamentos biosimilares ha conllevado un importante ahorro. Uno de los estudios más importantes desarrollados a nivel nacional fue el llevado a cabo por la Fundación Weber en 201729. Este análisis estimó un ahorro retrospectivo de 478 millones de euros entre 2009 y 2016, que se podrían ver incrementados en 1.965 millones de euros en el periodo 2017 a 2020, según su análisis prospectivo, con el uso de biosimilares.
En este sentido, cabe indicar que el marco regulatorio europeo está orientado a favorecer y a acelerar el acceso al mercado de los medicamentos biosimilares. El objetivo es fomentar la competencia en el mercado, contener el gasto farmacéutico y mejorar el acceso a medicamentos biológicos y a otros medicamentos innovadores.
• Switch de un medicamento biológico de referencia a un biosimilarEn relación al switch, el ECA de no inferioridad NOR-SWITCH, realizado en casi 500 pacientes con distintas enfermedades inmunomediadas, demostró que el cambio de IFX original al biosimilar CT-P13 no es inferior a un tratamiento continuado con el medicamento original en términos de eficacia, de seguridad e de inmunogenicidad30,31. En este ECA los pacientes fueron sometidos a un tratamiento estable con IFX durante al menos seis meses y, más adelante, aproximadamente la mitad de ellos cambiaron al biosimilar de IFX. Los datos desvelan que la eficacia y la seguridad fueron comparables entre grupos, obteniendo un margen de no inferioridad del 15%.
Los estudios observacionales publicados sobre el switch arrojan resultados similares en las distintas enfermedades inmunomediadas32,33.
Uso de medicamentos biosimilares en España• Barreras y facilitadores con el uso de biosimilaresUna encuesta publicada a nivel nacional concluyó que muchos profesionales se muestran relativamente cautelosos a la hora de utilizarlos en la práctica clínica4. Las principales barreras al uso de los medicamentos biosimilares encontradas en esta encuesta, al igual que en otras realizadas en países de nuestro entorno27,34-40, incluyen la falta de confianza, de conocimientos o de experiencia con el uso de estos medicamentos.
Junto con ello, la encuesta mostró la gran variabilidad a nivel hospitalario en la gestión de los medicamentos biosimilares (acceso, protocolos, etc.)4.
Por otro lado, los principales facilitadores para su uso son el acceso a la evidencia (ensayos clínicos y de datos empíricos de la vida real) sobre la disponibilidad, la eficacia, la seguridad y la intercambiabilidad, junto con la guía proporcionada por las sociedades científicas, la opinión de colegas de referencia y el desarrollo de protocolos locales4.
• Información para pacientesLa información al paciente y la toma de decisiones compartidas son fundamentales para favorecer la adherencia y evitar el efecto nocebo con el uso de medicamentos biosimilares41. Distintos estudios han puesto de manifiesto que para los pacientes sus médicos y otros profesionales de la salud, como enfermería y los farmacéuticos, son su principal fuente de información, y que desean estar implicados en las decisiones que atañen a su salud42,43.
En este sentido, con el uso de medicamentos biosimilares se recomienda explicar con detalle las características principales, la seguridad y la idoneidad del medicamento biosimilar, así como los cambios que se puedan decidir a lo largo de todo el tratamiento. Es recomendable también hacer partícipe a los pacientes del gasto farmacéutico y de la contribución de estos medicamentos para la sostenibilidad del sistema.
Actualmente se dispone de guías específicas para pacientes44,45, así como de material educativo (englobados en programas de soporte al paciente), servicio Home-Delivery, la tele-farmacia/tele-asistencia, o el asesoramiento en las asociaciones de pacientes.
• Research AgendaLa tabla 5 resume una propuesta de distintas posibles líneas de trabajo futuras con el uso de biosimilares.
Research Agenda con el uso de biosimilares
| # | Propuesta |
|---|---|
| 1 | Estudios a largo plazo en vida real en todas las indicaciones |
| 2 | Comité de biológicos en hospitales: estandarización de estructura, procedimientos, funciones, protocolos, etc. |
| 3 | Switch: definición de perfil de paciente |
| 4 | Desarrollo de programas educacionales para profesionales sanitarios |
| 5 | Desarrollo de programas de apoyo al paciente |
Con anterioridad al presente trabajo, nuestro grupo examinó la variabilidad del uso de biosimilares para el tratamiento de enfermedades inmunomediadas a nivel nacional4. En este estudio se constató que el nivel de conocimiento de los reumatólogos, dermatólogos, gastroenterólogos y farmacéuticos hospitalarios sobre las características fundamentales de los biosimilares y del marco normativo que los regula es insuficiente, especialmente teniendo en cuenta que la mayoría de los participantes llevan muchos años utilizando medicamentos biosimilares y tienen acceso a datos empíricos e información publicada por los órganos normativos7,46-54. Además, se constató una gran variabilidad en los hospitales españoles en cuanto a la gestión de estos medicamentos.
En este documento hemos descrito pormenorizadamente los aspectos más relevantes sobre los medicamentos biosimilares. Además, un grupo de expertos ha generado una serie de principios generales y recomendaciones, avaladas en un proceso Delphi, que pueden servir de marco para el uso de los medicamentos biosimilares. En este sentido queremos destacar varios de los mensajes generados. En primer lugar, comentar el reconocimiento a los medicamentos biosimilares por su contribución a la sostenibilidad del sistema y al acceso a terapias innovadoras, tal y como reflejan distintos estudios27-29. También los expertos destacan que los requerimientos de las agencias reguladoras para aprobar estos medicamentos son muy exigentes y rigurosos, lo que garantiza la seguridad de su uso en la práctica diaria. Actualmente los medicamentos biosimilares autorizados reúnen todas las características de calidad, eficacia y seguridad6,7,10. Además de la evidencia evaluada para su aprobación en las agencias reguladoras proveniente de ECA, se siguen publicando datos a medio y a largo plazo en vida real que avalan el uso de estos medicamentos17-26. Por ello, los expertos consideran imprescindible que los profesionales de la salud implicados en el uso de medicamentos biosimilares tengan un conocimiento actualizado y exhaustivo de las características de estos medicamentos, participen en estructuras y procesos relacionados con los mismos e informen pormenorizadamente a los pacientes.
Estamos convencidos de que este artículo contribuirá muy positivamente para mejorar el nivel de conocimiento y secundariamente del uso de medicamentos biosimilares. Pero también consideramos que se debe continuar trabajando en el ámbito de los medicamentos biosimilares para disminuir la variabilidad en la práctica clínica.
FinanciaciónEste proyecto ha sido financiado por una subvención irrestricta concedida por Fresenius Kabi España.
Contribución de los autoresEmilio Monte-Boquet contribuyó en el diseño del estudio, análisis e interpretación de los datos, revisó críticamente el artículo y aprobó la versión para su publicación. Ángeles Florez, Guillermo José Alcaín Martínez y Agustí Sellas participaron en el análisis y la interpretación de los datos, revisaron críticamente el artículo y aprobaron la versión para su publicación.
Conflicto de interesesGA ha participado en actividades de formación y asesoramiento con Fresenius, Nestlé, AbbVie, Janssen, Ferring, Pfizer, Tilots y Galapagos. AF ha realizado ensayos clínicos y actuado como conferenciante y consultora para AbbVie, Almirall, Amgen, Celgene, Janssen, Kyowa Kirin, Leo-Pharma, Lilly, Novartis, Pfizer, Roche Farma, Sanofi, Sun Pharma, Takeda y UCB Pharma. El resto de autores refieren no tener conflictos de intereses para esta publicación.
AgradecimientosNos gustaría dar las gracias a la Dra. Estíbaliz Loza por su ayuda con las tareas metodológicas y de estadística del estudio. También a los participantes del Delphi: José Luis Tandaipan Jaime, Nuria Rudi Sola, Conchita Pitarch Grau, Amparo Raga Beser, Mercedes Franco Donat, Jaime Poquet Jornet, Elvira Gea Rodríguez, Belén Hernández Muniesa, Arancha Pou Alonso, Rafael Belenguer Prieto, Jesús Sanchez Bursón, María Luisa Fernández Díaz, Daniel Carpio López, María Ángeles Castro Vida, Manel Velasco, José Manuel Mínguez Cortes, Montserrat Corteguera Coro, Javier García Miguel, Pedro Mas Morey, Marisa Gaspar, Juan José Alegre Sancho, Mireia Castillo Vilella, Isabel Betlloch Mas, Susana Antón González, Elena Matilla García, Santiago Sandoval, Francisco Castro Domínguez, Carlota Laura Iñiguez Ubiaga, José Rafael Úbeda Bonete, Raquel Moreno Díaz, Andrea García Guillén, Cristina Mata Arnaiz, José Ramón Blanch Comes, Luis Francisco Linares Ferrando, Maria Inmaculada Seguí Gregori, Juan Selva Otaolarruchi, Jesús Tornero Molina, Inmaculada López Rodríguez, Víctor Manuel López García, Coral Rivas Rivas, Miguel González Barcia, Teresa Abalde Pintos, Luis Antonio Pedraza Cezón, Marta Herrero Fernández, Beatriz del Pino Gaya, Paula Estrada, Tomás Ramón Vázquez Rodríguez, Regina Faré García, Antonio Vélez, Miguel Ángel Rodríguez Cabezas, Carmen Torres Martín, Francisco Javier Bécares Martínez, Juan Sánchez Bursón, Marta Martí Navarro, Lola Haro Martín, Elena Leonor Sirvent Alierta, José Manuel Martínez Sesmero, José Manuel Rodríguez Heredia, Marina Salido Olivares, Carlos Crespo Diz, Estefanía Moreno Ruzafa, Montserrat Rivero Tirado.


