



El avance en el conocimiento de las alteraciones bioquímicas que causan las enfermedades constitucionales óseas no tiene precedentes. La constatación de que su característica esencial es el trasfondo genético común a todas ellas ha dado lugar a una propuesta de alcance: sustituir el término «constitucionales» por «genéticas» para referirse a estas entidades. La comprensión de los mecanismos fisiopatológicos implicados, identificando el punto exacto de la vía metabólica alterada y sus sistemas de regulación y control, facilita realizar un diagnóstico preciso, basado en la colaboración interdisciplinar, en un tiempo muy inferior del que requería el enfoque tradicional. Además, aunque la correcta valoración de las manifestaciones clínicas y radiológicas sigue siendo crucial, el diagnóstico de certeza se basa cada vez con mayor frecuencia en la aplicación de las nuevas técnicas de análisis genético y molecular. Por último, el esclarecimiento de las complejas alteraciones subyacentes a estos trastornos descubre unas dianas moleculares de gran utilidad potencial en la investigación terapéutica de unas enfermedades que a menudo limitan de manera notable la calidad de vida y que, casi sin excepciones, todavía carecen de un tratamiento eficaz.
Recent years have seen an unprecedented increase in the knowledge and understanding of biochemical disturbances involved on constitutional bone disorders. Recognition of the genetic background as the common cause of these diseases prompted the substitution of the term «constitutional» by «genetic», in referring to them. Understanding physiopathological bases by finding out the altered metabolic pathways as well as their regulatory and control systems, favours an earlier and more accurate diagnosis based on interdisciplinary collaboration. Although clinical and radiological assessment remains crucial in the study of these disorders, ever more often the diagnosis is achieved by molecular and genetic analysis. Elucidation of the damaged underlying molecular mechanisms offers targets potentially useful for therapeutic research in these complex and often disabling diseases.
Las enfermedades constitucionales óseas (ECO) son un conjunto de trastornos, amplio y heterogéneo, cuya característica común es el fracaso en alguno de los sistemas celulares del hueso lo que da lugar a anomalías en la transformación del tejido óseo primitivo en hueso maduro1. Su expresión clínica y radiológica se puede manifestar por: alteraciones en el crecimiento total (disminución de talla o enanismo) o parcial (hipoplasia o aplasia), anomalías en el modelado (forma) del hueso, y alteraciones (por exceso o por defecto) en la densidad ósea1,2.
En el esquema clásico, las ECO —también denominadas enfermedades intrínsecas del hueso— se subdividían en dos grandes grupos: osteocondrodisplasias (displasias y distrofias) y malformaciones óseas (o disostosis). Aunque existe cierto grado de solapamiento entre ambas categorías, esta diferenciación, con variaciones a lo largo del tiempo, se ha considerado útil al menos desde el punto de vista teórico-conceptual2. Las «osteocondrodisplasias» son trastornos generalizados que se producen como resultado de una anomalía en la expresión génica de alguno de los tejidos básicos (óseo, cartilaginoso o conjuntivo) que conforman el esqueleto como estructura multifuncional. Ello condiciona que los fenotipos de estas entidades continúen evolucionando a lo largo de la vida de manera que huesos que en un principio están aparentemente sanos pueden mostrar alteraciones más tarde. Por su parte, las disostosis son anomalías en el número, el tamaño, la forma o la posición de un hueso individual (o en una combinación de ellos) que tienen su origen en una alteración en la blastogénesis, en las 6 primeras semanas del desarrollo embrionario. A diferencia de las osteocondrodisplasias, las malformaciones son localizadas y estáticas en el sentido de que no se extienden a estructuras no afectadas en origen, si bien pueden progresar en los huesos que ya estaban comprometidos al inicio.
Con el desarrollo y la aplicación de técnicas genéticas y bioquímicas complejas, cada vez más fiables e informativas, el concepto de ECO y la sistematización de las entidades que comprende ha ido evolucionando desde una visión basada en conceptos descriptivo-morfológicos a un enfoque más funcional que tiene como fondo el esclarecimiento de los mecanismos etiopatogénicos. De esta forma, las entidades que comprende este concepto nosológico pueden diferenciarse con mayor nitidez evitando la confusión que anteriormente existía. Nuestro objetivo es revisar los elementos clave que han contribuido a este cambio de indudable repercusión práctica por cuanto supone un marco muy distinto sobre el que establecer las bases del abordaje clínico y la investigación de estas enfermedades que, aunque individualmente son de baja frecuencia, en conjunto representan una parte no desdeñable de los cuidados sanitarios debido a la variedad de complicaciones ortopédicas y a la limitación en la calida de vida que ocasionan3,4.
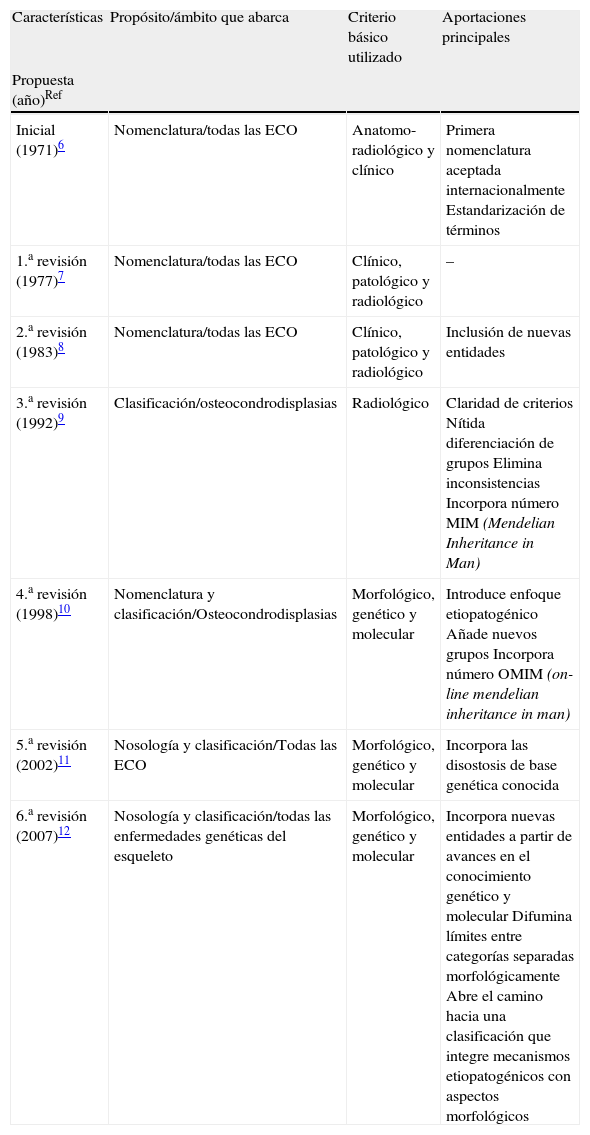
Perspectiva histórica de una difícil tarea: clasificar lo diverso y simplificar lo complejoAunque está fuera de nuestro propósito hacer una revisión exhaustiva de los sucesivos intentos de ordenar de manera sistemática las ECO, si nos parece necesario poner en perspectiva sus fundamentos y sus aportaciones principales. A mediados del siglo xx, el desarrollo de disciplinas como la radiología y la pediatría propiciaron una gran expansión del conocimiento de las enfermedades intrínsecas del esqueleto. Sin embargo, dada la complejidad de las estructuras óseas, el diverso origen de sus componentes y la heterogeneidad de los mecanismos fisiopatológicos subyacentes, desde el principio quedó clara la dificultad de identificar los trastornos que tenían verdadera entidad eliminando variantes sin fundamento (generalmente epónimos) y categorías superfluas. Tras varios intentos de sistematizar la taxonomía de las ECO, que fracasaron por utilizar enfoques inadecuados5, en 1969, un comité internacional de expertos elaboró la primera «Nomenclatura para las enfermedades constitucionales óseas» basada en criterios consistentes6. Siendo conscientes de las dificultades que esto suponía, los autores no pretendieron elaborar una clasificación general de las ECO. En realidad, su propósito era establecer categorías nosológicas bien definidas con una terminología homogénea que pudiera aceptarse con carácter universal, iniciando así un camino abierto a revisiones posteriores, que han sido seis hasta el momento presente7–12 (tabla 1).
Características y aportaciones principales de los sucesivos intentos de sistematizar la nomenclatura o clasificar las enfermedades constitucionales óseas.
| Características | Propósito/ámbito que abarca | Criterio básico utilizado | Aportaciones principales |
| Propuesta (año)Ref | |||
| Inicial (1971)6 | Nomenclatura/todas las ECO | Anatomo-radiológico y clínico | Primera nomenclatura aceptada internacionalmente Estandarización de términos |
| 1.a revisión (1977)7 | Nomenclatura/todas las ECO | Clínico, patológico y radiológico | – |
| 2.a revisión (1983)8 | Nomenclatura/todas las ECO | Clínico, patológico y radiológico | Inclusión de nuevas entidades |
| 3.a revisión (1992)9 | Clasificación/osteocondrodisplasias | Radiológico | Claridad de criterios Nítida diferenciación de grupos Elimina inconsistencias Incorpora número MIM (Mendelian Inheritance in Man) |
| 4.a revisión (1998)10 | Nomenclatura y clasificación/Osteocondrodisplasias | Morfológico, genético y molecular | Introduce enfoque etiopatogénico Añade nuevos grupos Incorpora número OMIM (on-line mendelian inheritance in man) |
| 5.a revisión (2002)11 | Nosología y clasificación/Todas las ECO | Morfológico, genético y molecular | Incorpora las disostosis de base genética conocida |
| 6.a revisión (2007)12 | Nosología y clasificación/todas las enfermedades genéticas del esqueleto | Morfológico, genético y molecular | Incorpora nuevas entidades a partir de avances en el conocimiento genético y molecular Difumina límites entre categorías separadas morfológicamente Abre el camino hacia una clasificación que integre mecanismos etiopatogénicos con aspectos morfológicos |
ECO: enfermedades constitucionales óseas.
Sin duda, el gran logro de la primera nomenclatura fue establecer una base sólida, evitando ideas equívocas y eliminando un buen número de términos y epónimos carentes de sentido. El sistema subdividía las «enfermedades constitucionales del hueso» —un total de 128 entidades sin contar las anomalías cromosómicas y las secundarias a enfermedades extraesqueléticas (grupos que no se desglosaron)— en 3 categorías principales: las de patogenia desconocida, las de patogenia conocida y las secundarias a alteraciones en sistemas extraesqueléticos (endocrinas hematológicas, etc.)6. Dentro de las de patogenia conocida, estaban las anomalías cromosómicas y las alteraciones metabólicas primarias (trastornos del metabolismo calcio-fósforo, mucopolisacaridosis, etc.). En cuanto a las de patogenia desconocida, claramente el apartado más amplio, incluía 4 grupos: osteocondrodisplasias, disostosis, osteólisis idiopáticas y alteraciones primarias del crecimiento (enanismo primordial, progeria y síndrome de Marfan, entre otras). Dentro de las osteocondrodisplasias —sin duda el subgrupo más nutrido—, consideraba: defectos del crecimiento de los huesos largos, de la columna o de ambos, trastornos por desarrollo desorganizado del cartílago y de los componentes fibrosos del esqueleto, y anomalías de la densidad del hueso. Por último, en el subgrupo de las disostosis se diferenciaba las de compromiso craneal y facial, las de predominio axial, y las de predominio en las extremidades.
Salvo por la inclusión de algunas nuevas entidades, la primera7 y la segunda8 revisiones, emitidas en 1977 y 1983, respectivamente (tabla 1) no aportaron cambios sustanciales. Con algunos matices, ambas mantenían los criterios morfológicos de la propuesta inicial así como el término «nomenclatura», y el ámbito de aplicación abarcaba al conjunto de las ECO. La tercera revisión, publicada en 1992, introducía una variación importante hablándose por primera vez de «clasificación» y focalizando su área de interés en las osteocondrodisplasias, grupo que aparecía notablemente expandido, en detrimento de las disostosis que el comité se reconocía incapaz de abordar con solvencia9. Además, la clasificación, con unas 200 entidades, se basaba en criterios radiológicos eliminado rasgos clínicos como la edad de comienzo y la historia natural debido a su variabilidad y, en ocasiones, inconsistencia. Por último, aunque incluía por primera vez el número MIM (mendelian inheritance in man, del catálogo de Mc Kusick) y alguna otra información genética, se reconoce que el conocimiento de las ECO era aún demasiado fragmentario para intentar una clasificación causal. Sin embargo, en la siguiente revisión10, publicada en 1998, la identificación de un buen número de los trastornos enzimáticos y celulares subyacentes permitió que por primera vez se elaborase una «nomenclatura y clasificación» con un enfoque causal. A pesar de los avances, esta cuarta revisión seguía limitándose casi en exclusiva al grupo de las osteocondrodisplasias, excluida la mayoría de las malformaciones óseas o disostosis que el grupo de expertos seguía considerándose incapaz de abordar. En esta propuesta, las categorías en la que inicialmente se subdividían las osteocondrodisplasias se sustituyeron por 32 grupos formados por entidades razonablemente homogéneas, con una diferenciación nítida entre ellos. No fue hasta la 5.a revisión11, emitida en 2001 y publicada un año más tarde, cuando la expansión de los conocimientos en esta área permitió volver a la concepción inicial, pasando a denominarse «Nosología y clasificación» y abarcando a todas las ECO al incluir de nuevo a las disostosis, en muchas de las cuales se había logrado localizar la región cromosómica involucrada e, incluso, sobre todo en el grupo de las craniosinostosis, la alteración genética causal.
Por fin, la 6.a revisión publicada en 200712 introduce una novedad de gran trascendencia al proponer que el término «genético» debe sustituir al de «constitucional» para denominar a estos procesos. Sin duda, este cambio, a la vez favorecido y exigido por los hallazgos genéticos y moleculares más recientes, es de gran alcance pues no sólo permite incorporar las nuevas entidades que se van reconociendo, sino que posibilita un reagrupamiento más dinámico y funcional de las mismas, acorde con la nueva información etiopatogénica que se va incorporando. En las siguientes líneas vamos a comentar las implicaciones de este último intento de ordenación sistemática de las ECO que abre las puertas a una clasificación que integre categorías morfológicos estructuradas sobre criterios consistentes con categorías relacionadas por su alteración patogénica-molecular de base.
Hacia una clasificación integralLa última revisión propuesta por el comité internacional de expertos (véase el extracto en la tabla 2) representa un cambio sustancial respecto a la clasificación anterior12. Los criterios que debe reunir una entidad para ser incluida son: a) afección esquelética relevante, acorde con la definición de displasia esquelética, enfermedad metabólica ósea, disostosis o síndrome de malformación y/o reducción; b) publicación o listado en el catálogo MIM; c) base genética probada o muy probable, y d) personalidad nosológica propia confirmada mediante análisis molecular o de ligamiento, o basada en la presencia de rasgos diagnósticos inequívocos o en la observación de múltiples casos individuales o familias. Utilizando esta combinación de criterios moleculares, bioquímicos y clinicorradiográficos se incluyen 372 entidades ubicadas en 37 grupos con límites bien definidos; de ellas, 215 enfermedades se asocian a diferentes alteraciones en uno o más de 140 genes.
Extracto de la última clasificación del Comité internacional: grupos y entidades principales que incluye.
| 1. Grupo FGFR3 (fibrodblast growth factor receptor 3): incluye la acondroplasia y trastornos relacionados |
| 2. Grupo del colágeno tipo II: acondrogénesis tipo 2, displasia espóndilo-epifisaria congénita… |
| 3. Grupo del colágeno tipo XI: incluye el síndrome de Stickler tipo 2 |
| 4. Grupo de las alteraciones de la sulfatación: incluye la acondrogénesis tipo 1B |
| 5. Grupo perlecan: incluye la displasia bisegmentada |
| 6. Grupo filamina: incluye el síndrome de Larsen (luxaciones congénitas múltiples) |
| 7. Grupo de la costilla-corta: displasia condro-ectodérmica… |
| 8. Displasias epifisarias múltiples y seudoacondroplasia |
| 9. Displasias metafisarias (tipo Schmid, McKusick y Jansen, entre otras) |
| 10. Displasias espondilometafisarias (tipo Kozlowski) |
| 11. Displasias espondiloepimetafisarias: con laxitud articular, inmunoósea, progresiva y seudorreumatoide |
| 12. Displasias espondilodisplásicas graves: acondrogénesis… |
| 13. Displasias espondilodisplásicas moderadas |
| 14. Displasias acromélicas: displasia trico-rino-falángica, artropatía familiar digital con braquidactilia… |
| 15. Displasias acromesomélicas |
| 16. Displasias meso y rizomélicas: discondrosteosis… |
| 17. Displasias con huesos curvados: displasia campomélica |
| 18. Displasias con huesos delgados |
| 19. Displasias con dislocaciones articulares múltiples: displasia de Desbuquois (tipo 1) |
| 20. Grupo de la condrodisplasia punctata |
| 21. Displasias osteoescleróticas neonatales: enfermedad de Caffey… |
| 22. Con ↑ de la densidad ósea: osteopetrosis (6 tipos), picnodisostosis, osteopoiquilia, meloreostosis… |
| 23. Con ↑ de la densidad y afectación meta o diafisaria: Camurati-Engelmann, hiperostosis endostal (Van Buchem), osteoectasia con hiperfosfatasia (Paget juvenil), enfermedad de Pyle, paquidermoperiostosis… |
| 24. Con ↓ de la densidad ósea: osteogénesis imperfecta (7 tipos), osteoporosis idiopática juvenil… |
| 25. Mineralización defectuosa: hipofosfatasia, raquitismos hipofosfatémicos, hipercalcemia hipocalciúrica… |
| 26. Trastornos del almacenamiento lisosomal: mucopolisacaridosis (10 tipos) |
| 27. Osteólisis: osteólisis expansiva familiar, progeria… |
| 28. Por desarrollo anómalo de componentes óseos: displasia fibrosa, exostosis cartilaginosas múltiples (3 tipos), encondromatosis (Ollier), fibrodisplasia osificante progresiva, heteroplasia ósea progresiva… |
| 29. Grupo de la displasia cleido-craneal |
| 30. Craniosinostosis y otras anomalías de la osificación craneal: síndromes de Pfeiffer, Apert, Crouzon.. |
| 31. Disostosis con predominio de afección cráneo-facial: síndrome oral-facial-digital… |
| 32. Disostosis con predominio de afección vertebral y costal |
| 33. Disostosis rotulianas: síndrome rótula-uña… |
| 34. Braquidactilias: osteodistrofia hereditaria de Albright… |
| 35. Hipoplasia de los miembros: anemia de Fanconi, síndrome Holt-Oram.. |
| 36. Polidactilia-sindactilia-trifalangismo |
| 37. Defectos de la formación articular: sinostosis múltiples (2 tipos), sinfalangismo proximal (2 tipos)… |
Los grupos 1 a 6 se han creado de nuevo o reformado en profundidad para dar cabida a entidades relacionadas por la alteración genética y molecular que las origina. En los grupos 7 a 16 prevalece el criterio de agrupamiento basado en la localización anatómica o en el patrón radiológico. Los grupos 17 a 19 se definen por criterios macroscópicos y por características clínicas (huesos curvados o adelgazados, presencia de dislocaciones múltiples). Los grupos 20 a 25 y 27 recogen las alteraciones en la mineralización y de la densidad ósea y, por tanto, prima el aspecto morfológico-radiológico. El grupo 26 representa el amplio grupo de los trastornos lisosómicos con afección esquelética. El grupo 28 incluye a las entidades asociadas al llamado «desarrollo anómalo de los componentes del esqueleto», como las exostosis, los encondromas y las diversas formas de calcificación ectópica. Finalmente, los grupos 29 a 37 (el grupo 29 incluye la displasia cleidocranial un ejemplo clásico de transición entre displasia y disostosis) se dedican al siempre difícil sector de las disostosis siguiendo criterios anatómicos (cráneo, cara, esqueleto axial, extremidades) con criterios adicionales que reflejan aspectos del desarrollo embrionario.
Tomando como referencia esta clasificación (que sigue en pleno proceso de mejora), la actual expansión de los conocimientos genéticos y bioquímicos permite reagrupar las ECO en función de su alteración patogénica-molecular en 7 categorías de claro significado funcional3,13. Reseñar en detalle, con arreglo a estas categorías, el elevado número de enfermedades cuya alteración genética ya ha sido identificada desbordaría los límites de esta revisión. Sin embargo, sí nos parece útil, a partir de los datos más recientes, señalar algunos ejemplos representativos dentro de cada grupo definido por el mecanismo patogénico involucrado (tabla 3).
Clasificación de las enfermedades constitucionales óseas de base genética conocida según su mecanismo patogénico-molecular: ejemplos más representativos.
| 1. Defectos en las proteínas estructurales extracelulares |
| Colágeno 1: osteogénesis imperfecta |
| Colágeno 2: acondrogénesis tipo 2, hipocondrogénesis, displasia espondiloepifisaria congénita, displasia espondiloepimetafisaria, displasia de Kniest, síndrome de Stickler tipo 1 |
| Colágeno 10: displasia metafisaria (tipo Schmid) |
| Proteína oligomérica de la matriz del cartílago (COMP): seudoacondroplasia, displasia epifisaria múltiple tipo 1 |
| Matrillin 3: displasia epifisaria múltiple tipo 3 |
| Perlecan: displasia bisegmentada y Schwartz-Jampel tipo 1 |
| Agrecano: displasia espondiloepisaria con artrosis primaria intensa (mutación en AGC 1) y displasia espondiloepimetafisaria recesiva (mutación en lectina tipo C) |
| 2. Defectos en las vías metabólicas |
| Fosfatasa alcalina (sin especificidad tisular): hipofosfatasia |
| ATPasa vacuolar (subunidad TCIRG-1): osteopetrosis grave infantil |
| Canal de cloro 7: osteopetrosis grave del adulto |
| Anhidrasa carbónica 2: osteopetrosis con calcificaciones intracraneales y acidosis tubular renal |
| Transportador de sulfato: acondrogénesis 1B, displasia diastrófica, displasia epifisaria múltiple recesiva |
| Arilsulfatasa E: condrodisplasia punctata ligada al cromosoma X |
| Receptor de potencial transitorio vaniloide 4 (TRPV 4): displasia espondilometafisaria tipo Kozlowski, braquiolmia y displasia metatrópica |
| 3. Defectos en el plegamiento o en la degradación de macromoléculas |
| Enzimas lisosómicas. Enfermedades por almacenamiento: mucopolisacaridosis |
| Catepsina K: picnodisostosis |
| Sedlin: displasia espondiloepifisaria tarda ligada al cromosoma X |
| 4. Defectos en hormonas y en mecanismos de transducción de señal |
| 25-α hidroxilasa de la vitamina D: raquitismo/osteomalacia vitamina D-dependiente tipo I |
| 1,25-α hidroxilasa de la vitamina D: raquitismo/osteomalacia resistente a la vitamina D |
| Proteína G estimuladora de la adenil-ciclasa (subunidad α) [GNAS1]: seudohipoparatiroidismo |
| Receptor de la PTH/PTHrp: displasia metafisaria tipo Jansen |
| Proteinasa del receptor de importación peroxisómica: raquitismo hipofosfatémico ligado al cromosoma X |
| Receptor del factor de crecimiento fibroblástico 23: raquitismo hipofosfatémico autosómico dominante |
| Receptor del factor de crecimiento fibroblástico 1: craniosinostosis (algunas formas de Pfeiffer) |
| Receptor del factor de crecimiento fibroblástico 2: craniosinostosis (Apert, Crouzon y otras variantes de Pfeiffer) |
| Receptor del factor de crecimiento fibroblástico 3: «familia de la acondroplasia»: displasia tanatofórica, acondroplasia y “SADDAN” (acondroplasia grave con acantosis nigricans) e hipocondroplasia |
| Activador del receptor del factor nuclear Kβ (TNFRSF11A): osteólisis expansiva familiar |
| 5. Defectos en proteínas nucleares y factores de transcripción |
| Gen SOX 9: displasia campomélica |
| Nucleotidasa 1 calcio activada: displasia de Desbuquois |
| 6. Defectos en oncogenes y en genes supresores de tumores |
| Exostosinas 1 y 2: exostosis múltiple tipo 1 y 2 |
| 7. Defectos en el procesamiento y metabolismo de ARN y ADN |
| Gen RMRP (codifica la parte de ARN de la MRP-RNAasa): hipoplasia cartílago-pelo |
Este grupo incluye algunas de las «familias» de ECO mejor caracterizadas y que afectan al colágeno más abundante en el hueso —el colágeno tipo 1— (también es importante en la piel y en los tendones), y al que predomina en el cartílago —el colágeno tipo 214. Por tanto, se trata del grupo quizá más numeroso y el que incluye varias de las entidades más frecuentes y que ocasionan mayor repercusión clínica. Entre ellas se encuentra la osteogénesis imperfecta, considerada el paradigma de las ECO que cursan con disminución de la densidad ósea. En 1979 Sillence propuso diferenciar 4 tipos de osteogénesis imperfecta: el tipo I, una forma común con escleróticas azuladas; el tipo II, letal en el periodo perinatal; el tipo III, una forma progresiva con escleróticas normales, y el tipo IV, similar al tipo I pero con escleróticas normales15. De esta forma esquemática de clasificar la osteogénesis imperfecta se ha pasado a incluir 7 tipos principales y 16 subtipos con alteraciones genéticas diferenciadas, lo que se traduce en distintos patrones de transmisión y notables diferencias clínicas12. Esta complejidad responde a la combinación de posibles mutaciones que pueden producirse en una u otra de las dos cadenas del colágeno tipo 1 (α-1 y α-2 con genes codificadores situados en diferentes cromosomas), de la «proteína asociada a cartílago», y de la prolil 3-1hidroxilasa (leprecan)16,17.
Por otra parte, las mutaciones en el colágeno tipo 2 (grupo 212) causan una gran variedad de ECO18. La más frecuente es la displasia espóndilo-epifisaria —congénita y tipo Strudwick—, que cursa con talla baja (con el tronco desproporcionadamente corto) displasia en múltiples epífisis articulares y platiespondilia. Otras mutaciones del colágeno tipo 2 dan lugar a formas leves de displasia espóndilo-epifisaria y a la displasia de Kniest (similar a la forma congénita de displasia espóndilo-epifisaria)19. Por último, pertenecen a este grupo la hipocondrogénesis y la acondrogénesis tipo 2, dos entidades relativamente frecuentes, letales antes del nacimiento4. En este grupo también cabe mencionar que mutaciones en la cadena α-1 del colágeno tipo 10 ocasionan la displasia metafisaria tipo Schmid20; la más frecuente y la menos intensa de las displasias metafisarias (grupo 9 de la última clasificación12). Dentro de las entidades que responden a este mecanismo también tiene interés considerar las alteraciones en dos proteínas relacionadas que sirven de puente entre proteínas de la matriz extracelular del cartílago: la proteína oligomérica de la matriz del cartílago (COMP por sus siglas en inglés) y la denominada «matrillin 3» (grupo 812). Las manifestaciones fenotípicas causadas por las mutaciones en estas proteínas dependen de la expresión tisular de los correspondientes genes. Así, las mutaciones en la COMP pueden dar lugar a una seudoacondroplasia o a la displasia epifisaria múltiple tipo 121,22, mientras las mutaciones de la matrillin 3 ocasionan la displasia epifisaria múltiple tipo 323. Cierran este capítulo las alteraciones en los proteoglucanos, cuya importancia en la matriz colágena los situaban como claros candidatos a engrosar la lista de proteínas defectuosas en relación con las ECO. Así hace ya varios años se demostró que mutaciones en uno de sus componentes, el perlecan, ocasionan la displasia disegmentada y el Schwartz-Jampel tipo 124. Más recientemente se han comunicado mutaciones en distintos genes relacionados con el agrecano (el proteoglucano más abundante de la matriz extracelular del cartílago y factor clave en la osificación endocondral, por lo que es determinante en la talla final) como causa de condrodisplasias que cursan con enanismo intenso y compromiso vertebral. En concreto, una mutación en la región variable del gen AGC 1 causa una variante de displasia espondiloepifisaria asociada a artrosis primaria intensa25. Por último, una forma recesiva de displasia espondiloepimetafisaria caracterizada por un enanismo muy intenso se debe a una mutación en el dominio «lectina tipo C»26.
Defectos en vías del metabolismo óseoLas entidades que responden a este mecanismo ponen de manifiesto la importancia de algunas vías metabólicas en el modelado y remodelado óseos. Así, distintas mutaciones en el gen de la fosfatasa alcalina, enzima clave del metabolismo del fosfato y del pirofosfato (elementos esenciales en el proceso de mineralización), ocasionan las distintas formas de hipofosfatasia: neonatal (letal), infantil y del adulto27. Por otra parte, mutaciones en los genes que codifican la síntesis de algunas de las proteínas implicadas en el proceso de transporte de H+ en el espacio de resorción (entre el borde «en cepillo» de los osteoclastos y la superficie ósea), donde la acidificación es un requisito indispensable para la disolución de los cristales de hidroxiapatita, son las responsables de distintas formas de osteopetrosis. Una mutación de la subunidad TCIRG 1 de la ATPasa vacuolar causa la forma grave infantil28,29, la relacionada con el canal de cloro 7 da lugar a la forma grave del adulto30, y la asociada con alteraciones de la anhidrasa carbónica 2 causa la osteopetrosis con calcificaciones intracraneales y acidosis tubular renal31. En cuanto a las enzimas del metabolismo del sulfato, las cuales se han involucrado como un factor importante en la osificación endocondral, se han descrito diversas mutaciones en el gen transportador de sulfato que dan lugar a distintas osteocondrodisplasias de herencia recesiva como: la acondrogénesis tipo 1B, la displasia diastrófica y la displasia epifisaria múltiple recesiva32. Además, una mutación en la arilsulfatasa E, una sulfatasa esteroidea que se ha observado en líneas celulares osteoblásticas y podría desempeñar un papel importante en la formación de cartílago, da lugar a la condrodisplasia punctata ligada al cromosoma X33. Pero el hallazgo más reciente y de mayor significado en este apartado se ha producido en relación con las displasias espondilometafisarias (incluye entidades de los grupos 10 y 1112), trastornos que, aunque con distinta intensidad y cierta variabilidad individual y de grupo, comparten características como talla baja, escoliosis (con platiespondilia y un aspecto peculiar de los pedículos vertebrales) y anomalías en las metáfisis de los huesos largos. El grupo incluye la displasia espondilometafisaria tipo Kozlowski (la entidad mejor definida), la braquiolmia (más leve) y la displasia metatrópica. El descubrimiento de distintas mutaciones alélicas en el gen que codifica el receptor vaniloide 4 de los canales de potencial transitorio (TRPV-4 por sus siglas en inglés) como causa de estos procesos34 es un buen ejemplo de cómo este nuevo enfoque basado en el conocimiento causal está modificando la concepción clínica y, por ende, el agrupamiento basado en rasgos morfológicos. El TRPV-4 es un miembro de la superfamilia de receptores iónicos de canal. En concreto es de una proteína del canal del calcio que se encuentra en las membranas celulares y que regula el flujo celular de este catión. Este hallazgo que afecta a tres entidades que comparten alteraciones fenotípicas similares prepara el camino para su agrupamiento en una nueva familia de displasias, lo que con toda probabilidad ocurrirá en la próxima revisión de la clasificación de las ECO.
Defectos en el plegamiento o en la degradación de las macromoléculasLa mayoría de las entidades incluidas en este apartado son enfermedades de almacenamiento lisosómico como las mucopolisacaridosis, ocasionadas por deficiencias en las enzimas de la degradación de los glucosaminoglucanos. Se trata de un amplio grupo de tesaurismosis con fenotipo de disostosis cuya descripción detallada (incluida la alteración genética correspondiente) fue incorporada a la Clasificación del Comité internacional de expertos a partir de la 5.a revisión (grupo 22)11 y ampliada en la última revisión (grupo 26)12. También forma parte de este mecanismo la picnodisostosis ocasionada por alteraciones en la catepsina K, una endoproteasa que actúa en la degradación de componentes de la matriz extracelular35. Por último, se debe mencionar que mutaciones en la Sedlin una proteína del retículo endoplásmico involucrada en el plegamiento y en el transporte de proteínas causa la displasia espondiloepifisaria tarda ligada al cromosoma X, la más común de las displasias espondiloepifisarias36.
Defectos en hormonas y en mecanismos de transducción de señalEste amplio, heterogéneo, y complejo grupo incluye entidades ocasionadas por trastornos en los mecanismos de señalización y comunicación celular que actúan a larga distancia (sistemas endocrinos), próximos al lugar donde se producen los medidores (paracrinos) e, incluso, en la propia célula donde se originan (autocrinos). Dentro de los trastornos endocrinos destacan las alteraciones en la síntesis de vitamina D y en el eje calcio-hormona paratiroidea (PTH). Así, mutaciones en el gen que regula la síntesis de la 25-α hidroxilasa dan lugar al raquitismo/osteomalacia vitamina D-dependiente tipo I37 y las de la 1,25-α hidroxilasa al raquitismo/osteomalacia resistente a la vitamina D38. También se ha demostrado que el seudohipoparatiroidismo se origina por mutaciones en la subunidad α de la proteína G estimuladora de la adenil-ciclasa, implicada la transducción de señal de la PTH39. Las alteraciones en el receptor de la PTH, con un importante efecto sobre la diferenciación de los condrocitos en la placa de crecimiento, ocasionan la displasia metafisaria tipo Jansen40. Por último, defectos en la proteinasa del receptor de importación peroxisómica causan un proceso relativamente frecuente: el raquitismo/osteomalacia hipofosfatémico ligado al cromosoma X41,42. Dentro de los trastornos asociados a alteraciones en los mecanismos de señal autocrino-paracrino, una forma menos frecuente de raquitismo/osteomalacia hipofosfatémica, la autosómica dominante, está ocasionada por una mutación en el receptor del factor de crecimiento fibroblástico 23, proteína de probada acción fosfatúrica43. En este mismo subgrupo destacan los trastornos del sistema de los factores de crecimiento fibroblástico, de gran importancia por estar involucrados en la proliferación celular, así como en el normal desarrollo y crecimiento de las extremidades y del área craneofacial44. Así, una mutación en el receptor del factor de crecimiento fibroblástico 1 da lugar a algunas formas de la craniosinostosis de Pfeiffer, mientras que las del receptor del factor de crecimiento fibroblástico 2 ocasionan las craniosinostosis de Apert y Crouzon, así como otras variantes de la de Pfeiffer45. Por su parte, distintas mutaciones en el receptor del factor de crecimiento fibroblástico 3, clave en la osificación endocondral y, por tanto, en la normal transformación de cartílago en hueso, dan lugar a los trastornos de la denominada «familia de la acondroplasia». Este importante grupo de entidades incluye, en orden decreciente de gravedad, la displasia tanatofórica, la acondroplasia y el «SADDAN» (acondroplasia grave con acantosis nigricans), así como la hipocondroplasia44,46–48. Por último, el receptor RANK, miembro de la superfamilia de receptores del TNF, tiene una influencia determinante en la diferenciación de los osteoclastos y en su respuesta a la PTH. Por ello, es lógico que mutaciones activadoras en el gen que codifica el activador del receptor del factor nuclear Kβ (TNFRSF11A) den lugar a la osteólisis expansiva familiar49.
Defectos en proteínas nucleares y factores de transcripciónLa característica básica de este grupo mayoritariamente constituido por disostosis es una alteración estructural en una proteína nuclear o factor de transcripción50. Por ejemplo, mutaciones en el gen SOX 9 que codifica a la proteína ligadora de ADN factor de transcripción tipo HMG, de gran importancia en la condrogénesis, están relacionadas con la displasia campomélica51. Otro ejemplo interesante de este grupo es la displasia de Desbuquois tipo 1, una condrodisplasia autosómica recesiva perteneciente al grupo de las dislocaciones múltiples (grupo 1912) que se caracteriza por una importante disminución de la talla (con acortamiento de las extremidades), laxitud articular y escoliosis progresiva. En esta entidad se han descrito 7 mutaciones distintas en el gen que codifica la nucleotidasa 1 calcio-activada, una enzima cuya función exacta se desconoce aunque alguno de sus sustratos se ha involucrado en importantes funciones de señalización, incluida la liberación de calcio intracelular52.
Defectos en oncogenes y en genes supresores de tumoresUn ejemplo representativo de este grupo que de momento incluye pocas entidades son las exostosis cartilaginosas tipos 1 y 2, ocasionados por mutaciones en los genes que codifican las exostosinas 1 y 2, implicadas en la diferenciación celular y en la génesis tumoral53.
Defectos en el procesamiento y metabolismo de ARN y ADNEste grupo se justifica sobre todo por las peculiaridades del gen RMRP (que codifica la parte de ARN de la MRP-ARNasa) cuya mutación da lugar a la hipoplasia cartílago-pelo54.
Epílogo: síntesis e implicacionesLos intentos iniciales de clasificar las ECO estaban basados en criterios parciales, faltos de uniformidad y carentes de una definición precisa. A los trastornos cuya existencia como entidad independiente era incierta se les solía denominar por epónimos que con frecuencia respondían a rasgos casuales o irrelevantes. Con el propósito de avanzar hacia una agrupación homogénea basada en criterios consistentes, un comité internacional de expertos elaboró en 1969 la primera «Nomenclatura para las enfermedades constitucionales óseas», la que fue aceptada con carácter universal. Desde entonces se han realizado 6 revisiones que además de establecer categorías nosológicas bien definidas y estandarizar la terminología han ido solventando problemas como el lugar que las disostosis, particularmente difíciles de encuadrar, deben ocupar. Pero lo más relevante ha sido la incorporación de los nuevos conocimientos de naturaleza genética y molecular. Así, la última revisión publicada en 2007, basada en la alteración patogénica de base en combinación con criterios morfológicos objetivos, abre las puertas a la clasificación del futuro, con grupos bien definidos de claro significado funcional.
Los nuevos hallazgos han puesto de manifiesto la extrema complejidad del hueso y del cartílago, con un amplio número de procesos celulares y vías metabólicas implicados en su estructura y funciones fisiológicas. En consecuencia, aunque las alteraciones clínicas y radiográficas siguen siendo esenciales para el diagnóstico diferencial de las ECO, el estudio genético y bioquímico ocupa ya un lugar destacado como herramienta para lograr un diagnóstico preciso. Además, este nuevo enfoque favorece el desarrollo de nuevas técnicas de diagnóstico y facilita la colaboración interdisciplinar, elementos cruciales en el abordaje de unos trastornos de gran complejidad. Por último descubre unas dianas terapéuticas que están permitiendo disponer de fármacos seguros y eficaces para tratar estas enfermedades, que más pronto que tarde deben perder su condición de «huérfanas».


