



El paciente, un hombre de 45 años de edad, ingresó a Emergencias debido a un padecimiento actual que inició 15 días antes de forma insidiosa y curso progresivo. Comenzó con debilidad simétrica y dolor en los pies y los tobillos, que se extendió hacia arriba hasta las rodillas. Más tarde, el dolor progresó a paraparesia, mostrando niveles de creatincinasa de 44.270 U/L e insuficiencia respiratoria que requirió ventilación mecánica. Se llevaron a cabo una electromiografía y un biopsia muscular del cuádriceps. El paciente respondió a la corticoterapia en pulsos y a manejo de soporte. La presentación de parálisis ascendente sugirió el diagnóstico de síndrome de Guillain-Barré; sin embargo, el grado de afectación de los músculos con rabdomiólisis explicó el daño neurológico por sí mismo. La biopsia reveló criterios patológicos para miopatía necrosante autoinmune (MNA), así como otros datos clínicos y de laboratorio. Además, reunió criterios para clasificarse como lupus eritematoso sistémico (LES). De acuerdo con la literatura revisada, este es el primer reporte de la asociación entre MNA y LES.
A 45 year-old man went to the emergency room due to disease duration of 15 days of insidious onset and progressive course. It began with symmetrical weakness and pain in feet and ankles that extends upward to the knees. Later, this progressed to paraparesis with Creatine phosphokinase levels of 44,270 U/L and respiratory failure that required mechanical ventilation. Electromyography and muscle biopsy of quadriceps were made. The patient responded to corticotherapy in pulses and supporting management. The presentation of ascending paresis suggested the diagnosis of Guillain-Barré syndrome. However, the degree of muscle involvement with rhabdomyolysis explains the neurological damage by itself. The biopsy revealed pathological criteria for necrotizing autoimmune miopathy (NAM), as well as other clinical and laboratory evidence. Patient disease continued and reached criteria for systemic lupus erythematosus (SLE). To our best knowledge, this is the first report of the NAM and SLE association.
Las miopatías inflamatorias constituyen un grupo heterogéneo de enfermedades musculares adquiridas subagudas, crónicas y raras veces agudas, que tienen en común la presencia de debilidad muscular proximal asociada a inflamación en la biopsia de músculo1. Debido a que estas enfermedades se presentan como el mayor grupo de miopatías adquiridas potencialmente tratables, el reconocimiento temprano es clínicamente fundamental2.
Las miopatías inflamatorias incluyen, clásicamente, la polimiositis, dermatomiositis, miositis esporádica por cuerpos de inclusión, el síndrome de sobreposición y la miositis no específica3. La incidencia de este grupo de enfermedades es de 1 por cada 100.000 habitantes4.
Las miopatías necrosantes, un subgrupo de miopatías inflamatorias5, con escaso componente inflamatorio, fueron reportadas por primera vez por Emslie-Smith y Engel6. En el 2004, el Muscle Study Group propuso una clasificación independiente para una entidad que se reconoce cada vez con mayor frecuencia, la miopatía autoinmune necrosante (NAM por sus siglas en inglés), de acuerdo con su presentación inmunopatológica, histológica y clínica3. Clínicamente, la NAM se presenta con debilidad simétrica de miembros superiores e inferiores de predominio proximal, de inicio agudo o subagudo, sumado a valores elevados de creatincinasa (CK) y hallazgos miopáticos en la electromiografía. En el análisis patológico, se observa ausencia de infiltrado inflamatorio prominente, con macrófagos en vez de células T como células efectoras1. En retrospectiva, se ha observado que una alta proporción de miopatías inflamatorias descritas como polimiositis fueron NAM7.
En la revisión de la literatura se hallan artículos de revisión y reportes de casos de enfermedad asociada a enfermedades autoinmunes, toxicidad por fármacos y malignidad3. Sin embargo, no se han encontrado estudios de prevalencia ni incidencia. El objetivo de este reporte de caso es dar a conocer una entidad clínica potencialmente tratable de presentación aguda, de tal forma que sea parte del diagnóstico diferencial inicial en estos casos.
Descripción del casoVarón de 45 años, natural y procedente de Lima, de ocupación vendedor ambulante, lo que implicaba deambulación intensa; acudió a sala de emergencias por cuadro de 15 dìas de evolución con debilidad muscular asociada a mialgias de aparición distal de miembros inferiores, parestesias de pies y manos, además de fiebre de 40°C en los primeros 3 días. El cuadro ascendió a las rodillas en los días siguientes, hasta que 10 días antes del ingreso las molestias se intensificaron. En ese lapso, acude a centro de salud donde le indicaron antiinflamatorios no esteroideos y relajantes musculares. En el momento del ingreso, el dolor y la debilidad fueron de predominio proximal, tanto en miembros superiores e inferiores, llegando a imposibilitar la bipedestación.
Dentro de antecedentes patológicos, presentó alergia a las penicilinas, fiebre tifoidea a los 14 años, litiasis renal a los 15 años e infección por dengue a los 32 años. Diagnosticado de hipertensión arterial en tratamiento irregular con captopril 25mg; otros fármacos ingeridos fueron esteroides anabólicos semanalmente durante las 6 semanas previas al ingreso. Como hábitos nocivos refirió consumo de tabaco y alcohol esporádico. Entre sus antecedentes familiares de importancia, una tía materna falleció por cáncer cuello uterino.
En sus funciones biológicas resaltó constipación desde 6 días antes del ingreso. En el examen físico: funciones vitales, frecuencia cardiaca 100 latidos por minuto, frecuencia respiratoria 28 ventilaciones por minuto, presión arterial 90/60mmHg y temperatura 37,2°C. El paciente se encontraba diaforético y presentaba dolor al tacto en los miembros inferiores. Sensibilidad conservada. Con respecto al aparato locomotor, la motilidad pasiva presentó tono muscular disminuido, sin resistencia al movimiento. En la motilidad activa presentó fuerza distal y proximal en los miembros inferiores de 2/5 y en miembros superiores 3/5. En el examen del sistema nervioso, los reflejos osteotendinosos bicipital, rotuliano y aquíleo se valoraron en++++ simétricamente; la evaluación de los pares craneales no mostró anormalidades.
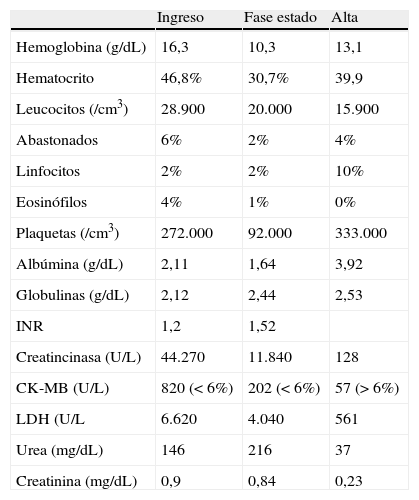
Los exámenes de laboratorio de ingreso indicaron leucocitosis, linfopenia, CK 44.270 U/L (tabla 1). En el examen de orina, presentó más de 100 hematíes y 10-20 leucocitos por campo, proteínas ++/+++ y sangre oculta +++/+++. Infectología: descartó etiología viral o bacteriana (VIH, test de serología luética, HTLV-I, HBV, virus de la hepatitis C, citomegalovirus, virus de Epstein-Barr, Cryptoccocus sp.). Asimismo, el servicio de Toxicología descartó asociación del cuadro clínico a etiología tóxica.
Principales variables de laboratorio
| Ingreso | Fase estado | Alta | |
| Hemoglobina (g/dL) | 16,3 | 10,3 | 13,1 |
| Hematocrito | 46,8% | 30,7% | 39,9 |
| Leucocitos (/cm3) | 28.900 | 20.000 | 15.900 |
| Abastonados | 6% | 2% | 4% |
| Linfocitos | 2% | 2% | 10% |
| Eosinófilos | 4% | 1% | 0% |
| Plaquetas (/cm3) | 272.000 | 92.000 | 333.000 |
| Albúmina (g/dL) | 2,11 | 1,64 | 3,92 |
| Globulinas (g/dL) | 2,12 | 2,44 | 2,53 |
| INR | 1,2 | 1,52 | |
| Creatincinasa (U/L) | 44.270 | 11.840 | 128 |
| CK-MB (U/L) | 820 (< 6%) | 202 (< 6%) | 57 (> 6%) |
| LDH (U/L | 6.620 | 4.040 | 561 |
| Urea (mg/dL) | 146 | 216 | 37 |
| Creatinina (mg/dL) | 0,9 | 0,84 | 0,23 |
CK-MB: creatinincinasa fracción MB; INR: razón normalizada internacional; LDH: lactato deshidrogenasa.
La electromiografía informó patrón mixto neurógeno-miógeno, no actividad espontánea anormal del tipo denervación y fibrilaciones. Se consideró probable polirradiculitis inflamatoria aguda a predominio desmielinizante, a confirmar por estudio de líquido cefalorraquídeo. El líquido cefalorraquídeo informó no disociación albúmino-citológica; recuento celular 5, polimorfonucleares 10%, mononucleares 10%, glucosa 83mg/dL y proteínas 26mg/dL.
Por todo lo anterior, se planteó parálisis fláccida con sospecha de etiología autoinmune, tóxica, metabólica o neoplásica; insuficiencia respiratoria aguda tipo oxigenatoria por probable neumonía, rabdomiólisis con sospecha de etiología tóxica o inflamatoria, síndrome inflamatorio de respuesta sistémica, alcalosis respiratoria compensada, hiponatremia, hipocalcemia e hiperlactatemia.
Durante la evolución, el cuadro respiratorio empeoró en los siguientes días, los hallazgos de laboratorio se interpretaron como insuficiencia respiratoria (oxigenatoria y ventilatoria), acidosis metabólica asociada a acidosis respiratoria y azoemia; además, presentó Glasgow 10, por lo que requirió soporte ventilatorio mecánico. Le iniciaron corticoterapia con pulsos de metilprednisolona de 1.000mg/24h por vía intravenosa, uno diario por 3 días, por sospecha de etiología autoinmune.
En orden cronológico desde el quinto hasta el decimosexto día de hospitalización, se obtuvo anti-ADNds positivo (título 1/160), presentó cuadro psicótico paranoide, con alucinaciones predominantemente visuales y de vacío corporal, proteinuria de 1.000mg/dL, convulsiones, efusión pleural laminar bilateral mediante ecografía y trombocitopenia de hasta 25.000 U/cc. Con todos estos criterios, se planteó el diagnóstico de lupus eritematoso sistémico (LES) activo8.
El informe de la biopsia de músculo vasto externo del cuádriceps indicó presencia de infiltrado inflamatorio con macrófagos entre las fibras musculares destruidas y ausencia de vasculitis, hallazgos anatomopatológicos compatibles con miopatía necrosante (figs. 1 y 2).


Se cambió la vía de administración de corticoterapia a vía oral de prednisona 70mg/día, además del manejo de soporte con antibióticos, anticoagulantes, protectores gástricos y manejo del balance hidroelectrolítico. Se disminuyó la dosis de prednisona progresivamente. Se le retiró la intubación endotraqueal 3 semanas después del ingreso; días después salió de cuidados intensivos e ingresa a pabellón de Medicina Interna con valores normalizados de gases arteriales y hemograma Hb 10,6/L, Hto 27,8 L/L, leucocitos 10.390/cm3, abastonados 0%, linfocitos 9%, plaquetas 410.000, valores de CK 3.77 U/L, urea 42mg/dL y creatinina 0,4mg/dL. El sedimento urinario, con 8-10 leucocitos y 2-4 hematíes por campo, proteínas y sangre oculta negativo. En el pabellón, los cuadros respiratorio y hematológico evolucionaron favorablemente. Se disminuyó la dosis de prednisona a 60mg/día vía oral. La fuerza de miembros superiores e inferiores continuaba comprometida de 2/5 y 3/5, respectivamente. Oncología descarta etiología neoplásica (alfafetoproteína, antígeno carcinoembrionario, Ca19.9, Ca72.4, antígeno prostático específico, B2-microglobulina, negativos).
Fue dado de alta con indicaciones de seguir terapia física y rehabilitación, prednisona 50mg/día y control por consultorio de Medicina Interna. Nueve meses después de su alta, el paciente descontinuó sus visitas al médico del hospital, pero continúa con prednisona 20mg/día, calcio-vitamina D y pregabalina 75mg para el manejo del dolor, indicados por un médico de otro centro de salud. A la fecha de la redacción de la presente, realiza ejercicios de terapia física en su domicilio y camina con la ayuda de un bastón.
DiscusiónEl análisis inicial del cuadro indicó el diagnóstico de síndrome de Guillain-Barré durante gran parte de los primeros días, dadas las características clínicas que presentaba el paciente, como la presencia de debilidad muscular distal que empezó de manera simétrica en miembros inferiores, de progresión ascendente hasta insuficiencia respiratoria y necesidad de ventilación mecánica. Sin embargo, las parestesias de pies y manos deben valorarse con cautela en el momento de considerar los criterios clínicos, puesto que cuando son síntomas del síndrome de Guillain-Barré estas suelen preceder a la debilidad y no aparecer concomitantemente a esta, como en este caso9. Además, la presencia de fiebre alta durante 3 de los primeros días obliga a pensar en otros probables diagnósticos y a poner en duda el de Guillain-Barré10. Así mismo, la favorable respuesta a la corticoterapia aleja aún más el diagnóstico inicial de síndrome Guillain-Barré y apoya al de enfermedades autoinmunes11,12.
Las cifras elevadas de CK y deshidrogenasa láctica fueron evidencia de destrucción muscular y, sumadas al edema de los miembros afectados, hizo necesario plantear como principal diagnóstico el de miositis; la rápida progresión e intensidad y los valores de laboratorio exigieron pensar en la naturaleza necrotizante de la misma.
La ingesta de esteroides anabólicos ha sido reportada como una de las posibles causas de cuadros de rabdomiólisis en varias publicaciones13,14. El ejercicio intenso, factor presente en nuestro caso debido a las jornadas laborales del paciente, también ha sido reportado como un posible detonante del cuadro15,16.
El cuadro clínico y el hallazgo anatomopatológico del caso presentado se explican y fueron compatibles con miopatía inflamatoria del tipo autoinmune necrosante. La NAM se distingue por la ausencia de infiltrado inflamatorio prominente y macrófagos como células efectoras en vez de linfocitos T; además, la inmunohistoquímica no suele evidenciar expresión de MHC-I, como en polimiositis1,6,17. Generalmente, se caracteriza clínicamente por un inicio subagudo de debilidad de miembros proximal y simétrica, niveles elevados de CK y hallazgos de miopatía en la electromiografía; asimismo, por su respuesta a altas dosis de corticoides2,18. Sin embargo, en el caso presentado, el inicio fue distal y la progresión, proximal. Se ha encontrado que la NAM ha sido asociada con desórdenes del tejido conectivo, infecciones virales, fármacos, en particular los inhibidores de reductasa HMG-CoA o estatinas19, y malignidad3,20. Se ha descrito el anticuerpo anti-SRP como marcador de la NAM, en especial relacionado con estatinas; sin embargo, no se cuenta aún con un kit comercial en nuestro medio para su evaluación21.
La anamnesis descartó el uso de estatinas u otra entidad tóxica asociada a NAM; los exámenes de laboratorio no hallaron antígenos virales positivos como VIH, HTLV o HBV, que también pueden estar asociados3, y la respuesta al tratamiento y el seguimiento por 9 meses aleja tanto la etiología neoplásica como tóxica10, por lo que se colige que el paciente presentó una enfermedad autoinmune desencadenada por el consumo de esteroides anabólicos, el ejercicio físico excesivo junto con la exposición solar, sumado a alguna susceptibilidad genética no reportada en los antecedentes familiares.
Durante el curso de su enfermedad, el paciente presentó más de 4 criterios de clasificación para LES, lo cual no excluye el diagnóstico de NAM, sino que podría llegar a considerarse un síndrome de sobreposición. No se usaron agentes citotóxicos, como azatioprina, ciclofosfamida o micofenolato de mofetilo; a pesar de que las guías de práctica clínica para LES señalan que estos agentes previenen las recurrencias a largo plazo, numerosos pacientes responden al tratamiento solo con corticoides, como ocurre en este caso8,22. El LES podría explicar el cuadro neurológico que indicó en un inicio un síndrome de Guillain-Barré, asociación reportada en la literatura10,23,24. Sin embargo, en el paciente no hubo disociación albúmino-citológica ni algunas características clínicas discutidas previamente.
Asimismo, la literatura reporta LES asociado a compromiso muscular variable que puede llegar a la rabdomiólisis25,26, casos en los cuales es indudable el antecedente de infección o clara reacción a algún fármaco, así como miopatías inflamatorias27-29. Sin embargo, no se ha hallado en la literatura la asociación presentada en este paciente de miopatía inflamatoria tipo NAM, de presentación inusualmente distal, y LES, lo que resalta la importancia de comunicar este caso. Asimismo, ante un cuadro de inicio similar, debe integrarse prontamente esta entidad al ejercicio clínico, dado que pone en riesgo la vida del paciente y requiere manejo multidisciplinario.
Responsabilidades éticasProtección de personas y animalesLos autores declaran que para esta investigación no se han realizado experimentos en seres humanos ni en animales.
Confidencialidad de los datosLos autores declaran que han seguido los protocolos de su centro de trabajo sobre la publicación de datos de pacientes y que todos los pacientes incluidos en el estudio han recibido información suficiente y han dado su consentimiento informado por escrito para participar en dicho estudio.
Derecho a la privacidad y consentimiento informadoLos autores han obtenido el consentimiento informado de los pacientes y/o sujetos referidos en el artículo. Este documento obra en poder del autor de correspondencia.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.
Se agradece al médico patólogo César Chian García por las descripciones anatomopatológicas de las láminas del paciente. Así mismo, hacemos extenso el agradecimiento al médico neurólogo Luis Torres Ramírez por las precisiones en las manifestaciones neurológicas del cuadro y la revisión del manuscrito.


