




La resistencia a la acción de la insulina es una situación clínica que suele acompañar a las enfermedades inflamatorias crónicas como la artritis reumatoide. Su presencia puede aumentar la morbi-mortalidad de las enfermedades a las que se asocia. En qué consiste, cuál es su etiopatogenia, cómo se mide y qué implicaciones clínicas y terapéuticas tiene en pacientes con artritis reumatoide es un tema que no es familiar para reumatólogos y que abordamos en esta revisión.
Resistance to insulin action is a feature that accompanies certain diseases among which chronic inflammatory states like rheumatoid arthritis are included. What is, what its pathogenesis is, how it is measured, and what clinical and therapeutic implications have in rheumatoid arthritis patients is a topic not familiar to rheumatologists that is reviewed in this paper.
La insulina es una hormona polipeptídica producida y segregada por las células beta de los islotes de Langerhans del páncreas en forma de precursor inactivo llamado proinsulina. Interviene en el aprovechamiento metabólico de los nutrientes, sobre todo en el anabolismo de los carbohidratos y su acción metabólica es la de mantener la homeostasis de la glucosa así como promover una utilización eficiente de esta. Su efecto máximo lo define la capacidad de respuesta de esta, mientras que la concentración de insulina necesaria para llevar a cabo la mitad de la respuesta máxima se define como sensibilidad a la insulina.
La resistencia insulínica (RI) es un estado en el que una concentración alta de insulina se relaciona con una respuesta inadecuada de la glucosa con niveles normales o altos de glucemia. Es pues, una situación patológica caracterizada por la pérdida de la respuesta fisiológica de los tejidos periféricos a la acción de la insulina (endógena o exógena). La existencia de la RI se conoce desde el uso comercial de la insulina, e inicialmente se refería a pacientes que necesitaban dosis altas de insulina para controlar su hiperglucemia. Este hecho se debía a la presencia de anticuerpos como consecuencia del uso de insulinas no humanas. La desaparición de estos anticuerpos con el uso de la insulina recombinante humana ha cambiado la visión de la patogenia de la RI. El espectro clínico que acompaña a la RI es muy variado y viene dado por cómo actúa la insulina en diferentes tejidos dianas. Dado que esta hormona promueve la toma de glucosa en el músculo esquelético mediante la estimulación de GLUT4 (glucose transporter type 4), la RI ocasiona que esta señal se produzca de forma defectuosa, ocasionando un descenso en la captación de glucosa por el músculo. En el hígado, la insulina, de forma fisiológica, inhibe la expresión de enzimas gluconeogénicas por lo cual, en un estado de RI aumenta la síntesis hepática de glucosa. En lo que se refiere al tejido adiposo, la insulina desciende la actividad de determinadas lipasas lo que ocasiona un descenso en la producción de ácidos grasos libres: efecto antilipolítico. Por tanto, en un estado de RI, tendrá lugar un aumento de ácidos grasos libres. Lo más frecuente en la RI es la presencia de hiperglucemia en presencia de dosis altas de insulina, pero no es raro encontrar pacientes con normoglucemia con alteraciones tales como acantosis nigricans, hiperandrogenismo, amenorrea e infertilidad, síndrome de ovario poliquístico, obesidad, alopecia, hirsutismo, hipertrigliceridemia, etc.
Hoy en día se considera a la RI como un componente de varias enfermedades con etiologías y manifestaciones de diferentes tipos que se enumeran en la tabla 1. Parece clara la relación entre RI y disfunción microvascular como manifestación precoz de aterosclerosis1. Para algunos autores todas estas manifestaciones clínicas se agrupan en un mismo síndrome llamado síndrome X o síndrome metabólico en el cual la RI sería un denominador común y el nexo de unión de todas ellas, sin embargo, según el NCEP-ATPIII (National Cholesterol Education Program-Adult Treatment Panel III)2 no es indispensable la presencia de RI para el diagnóstico de síndrome metabólico.
Causas y manifestaciones clínicas de la resistencia insulínica
| Causas genéticas |
| Leprechaunism, síndrome de Rabson-Mendenhall (mutaciones del receptor de la insulina) Diabetes mellitus tipo 2 Lipodistrofias |
| Formas secundarias |
| Obesidad, síndrome metabólicio, síndrome de ovario poliquístico |
| Exceso de hormonas contrarreguladoras (glucorticoides, lactógeno placentario, hormona del crecimiento) |
| Inactividad |
| Infección, inflamación |
| Anticuerpos anti-insulina |
| Miscelanea (uremia, cirrosis, cetoacidosis) Manifestaciones clínicas |
| Homoestasis de la glucosa |
| Variable, desde diabetes franca a glucemia alterada en ayunas, euglucemia e hipoglucemia |
| Manifestaciones clínicas |
| Acantosis nigricans, alopecia |
| Aparato reproductor |
| Amenorrea, hirsutismo, virilización, infertilidad |
| Tejido adiposo |
| Variable, desde normal a lipoatrofia, lipohipertrofia y besidad |
| Hipertrigliceridemia |
| Musculoesquelético |
| Calambres, hipertrofia muscular, pseudoacromegalia |
Existe un debate acerca de cómo se debe medir la RI ya que son varios los métodos útiles. A continuación se resumen de forma abreviada las diferentes técnicas e índices disponibles dentro de los cuales tenemos métodos directos, métodos indirectos e índices simples de sustitución3.
Métodos directos- •
Clamp (pinzamiento) hiperinsulinémico euglucémico. Se acepta como el método de referencia para determinar la sensibilidad a la insulina en humanos. Tras el ayuno nocturno, se administra insulina endovenosa a una tasa por minuto dependiente de la superficie corporal. Esta infusión de insulina lleva a un nivel sanguíneo de insulina que está por encima del habitual (hiperinsulínico). Como consecuencia de esto, el gasto de glucosa en músculo esquelético y grasa aumenta, mientras que por el contrario, la producción hepática de glucosa es inhibida. Bajo estas condiciones se comienza la administración de dextrosa al 20% para mantener (clamp) la glucosa en nivel normales. Después de varias horas de constante perfusión de insulina, se alcanzan condiciones de equilibrio general, para insulina plasmática, glucosa en sangre, y tasa de infusión de dextrosa. Suponiendo que el estado de hiperinsulinemia es suficiente para suprimir totalmente la producción hepática de glucosa, y puesto que no hay ningún cambio neto en las concentraciones de glucosa en sangre bajo estas condiciones, la tasa de infusión de glucosa debe ser igual a la tasa de utilización de glucosa. De esta forma se puede calcular la sensibilidad a la insulina, bajo la ecuación:
- •
Test de supresión de insulina. Tras ayuno de 8h, se administra una dosis de somatostatina para inhibir la secreción endógena de insulina y glucagón. Posteriormente, se administra insulina (25mU·m−2·min−1) y glucosa (240mg·m−2·min−1) durante 3h en una misma vía venosa mientras que en otra se determinan periódicamente glucosa e insulina. A las 2h aproximadamente se obtienen unas concentraciones de glucosa e insulina en estado de equilibrio que serán mayores en sujetos insulinorresistentes y más baja en sujetos insulinosensibles. Tiene la ventaja, frente al Clamp que técnicamente es menos exigente y que la concentración de equilibrio entre glucosa e insulina se obtienen de forma más precoz.

- •
Modelo de análisis mínimo tras mediciones frecuentes en test de tolerancia a la glucosa endovenosa o FSIVGTT (frequently sampled intravenous glucose tolerance test). Después del ayuno nocturno, se administra un bolo de 0,3g/kg de glucosa y, tras 20min, se administra otro bolo de 4mU/kg/min de insulina durante 5min. Tras esto se toman determinaciones sanguíneas de glucosa e insulina cada minuto durante 10min, luego cada 3min hasta los 30min y de ahí en adelante cada 10min hasta el minuto 180. Los datos obtenidos son procesados mediante un programa informático diseñado a este fin (MINMOD, minimal model software) para determinar la sensibilidad a la insulina. Con respecto a los dos métodos directos descritos previamente, tiene la diferencia de que usa datos dinámicos, no de concentraciones en equilibrio y que técnicamente es más fácil y reproducible.
- •
Test de tolerancia oral a la glucosa (TOG). Test ampliamente utilizado para diagnosticar la intolerancia a la glucosa o la diabetes tipo 2. Tras ayuno nocturno, y toma de 75g de glucosa, se determina la glucemia a los 0, 30, 60 y 120min. Reflejaría la eficacia que tiene el organismo para disponer de la glucosa tras una sobrecarga o comida. Imitaría la dinámica de la glucosa e insulina de forma más fisiológica que el clamp o el FSIVGTT, pero tiene el inconveniente obvio de que tolerancia a la glucosa y sensibilidad a la insulina no son conceptos equivalentes.
Índices simples de sustitución. Se asume que en ayunas, la glucosa y la insulina están en equilibrio homeostático y que expresan una secreción hepática de glucosa constante, es decir, la insulina secretada por la célula β determina un nivel de insulina relativamente constante que será mayor o menor de conformidad con la resistencia/sensibilidad de esta, de tal manera que la producción hepática de glucosa concordará con el gasto global de glucosa en condiciones de ayuno. Estos índices que usan solo glucemia e insulina plasmática son herramientas menos costosas que se pueden utilizar en estudios epidemiológicos, ensayos clínicos y práctica clínica. Los 4 primeros que se mencionan a continuación: 1/insulina, ratio glucosa/insulina Homeostasis model assessment (HOMA) y Quantitative insulin sensitivity check index (QUICKI) son índices simples de sustitución de métodos indirectos, los restantes derivan de modelos dinámicos.
- •
1/insulinemia en ayunas. En sujetos sanos la elevación de la insulina se corresponde con una mayor resistencia a esta, por tanto, 1/insulina sería un índice que disminuye a medida que el paciente se hace insulinorresistente. Tiene el inconveniente de que la insulinemia no se distribuye de forma normal, por lo cual no existe una correlación lineal fuerte entre este índice y el método clamp. Del mismo modo, este índice tampoco tiene en cuenta la baja secreción de insulina que tiene lugar en estadios finales de la hiperglucemia en los pacientes con diabetes o con intolerancia a la glucosa, por lo cual, no sería conveniente su uso en estos pacientes con reserva pancreática deteriorada.
- •
Ratio glucosa/insulina. Este ratio en pacientes no diabéticos sería equivalente al índice 1/insulina. Tiene el inconveniente de que una misma insulinemia puede corresponderse con diferentes glucemias en función de si el sujeto es diabético o no. Se considera un índice conceptualmente erróneo de sensibilidad a la insulina.
- •
Modelo de evaluación de la homeostasis (HOMA). Descrito en 19854, es un modelo que usa la interacción dinámica entre glucosa e insulina para predecir la sensibilidad a la insulina o la producción de la célula β. Asume que existe un bucle de retroalimentación positivos entre el hígado y la célula pancreática, es decir, la concentración de glucosa la define la producción hepática que está inhibida por la insulina, mientras que la insulinemia es el reflejo de la respuesta de la célula β a la concentración de glucosa. Por tanto, la resistencia a la insulina vendría dada por una respuesta disminuida de la producción hepática de glucosa al efecto inhibidor de la insulina. Este modelo en su forma actualizada (http://www.dtu.ox.ac.uk/homa), HOMA2, permite determinar la sensibilidad a la insulina (%S) y la función de la célula beta (%B). Posee una razonable correlación con el método clamp, es útil en pacientes con diabetes mellitus moderada y estados de insulinorresistencia asociados a otras enfermedades, pero no es aplicable en pacientes con función pancreática muy dañada o nula.
- •
Índice de verificación cuantitativo de sensibilidad a la insulina (QUICKI). Al igual que el HOMA representa un modelo matemático de la relación entre insulina y glucosa, pero tiene un origen diferente. Se basa en que los análisis de sensibilidad de los datos de insulina y glucosa durante los primeros 20min del FSIVGTT contienen información crítica sobre la sensibilidad a la insulina. En estos datos, la insulinemia en ayunas no sigue una distribución normal pero su transformación logarítmica mejora la correlación de esta con el método clamp. A tal fin, la fórmula: QU1CK1=1/log insulina+log glucosa tiene una correlación óptima con el método clamp que se mantiene en pacientes diabéticos o con una función β disminuida. Tiene la ventaja sobre HOMA que se correlaciona mejor con el método clamp en paciente diabéticos y obesos.
- •
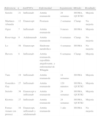
Índice de Matsuda. Representa una combinación que refleja tanto la sensibilidad insulínica hepática como muscular mediante la fórmula:
Se correlaciona de forma razonable con el clamp.
- •
Índice de Gutt. Al igual que el de Matsuda, es un test derivado de técnicas dinámicas que guarda estrecha correlación con el método clamp pero de compleja determinación:
Donde G0 es glucosa en ayunas, G120 glucosa a los 120min.
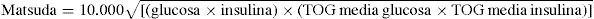
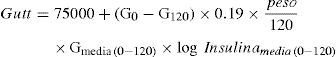
Desde los años 50 se ha venido observando una cierta correlación entre los estados inflamatorios crónicos y la resistencia a la acción de la insulínica. En las últimas décadas se ha puesto de manifiesto la relación entre obesidad e inflamación, procesos ambos claramente ligados con RI. Estudios en obesidad, en pacientes con diabetes mellitus tipo 2 y en otros estados relacionados con RI han encontrado niveles elevados de factor de necrosis tumoral (TNFα), interleuquina IL-6 e IL-85–7. También es habitual en estos estados la elevación de la proteína C reactiva, un reactante de fase aguda no especifico. Se ha observado la existencia de un estado de inflamación de baja intensidad en relación con obesidad sin que se conozcan a fondo los mecanismos que lo generan. Una teoría sería que la expansión del tejido adiposo (hipertrofia e hiperplasia de adipocitos) lleva a hipoxia celular local y a la activación de vías de señalización inflamatorias. También se sabe que determinadas citoquinas secretadas por los adipocitos, adipoquinas, como la resistina, adiponectina o leptina tienen acciones proinflamatorias y se relacionan con estados de RI8. De igual manera, la infiltración grasa por el hígado produce inflamación local en este órgano lo cual podría llevar a activación de las células de Kupffer que también favorecen estados de insulinorresistencia. Y por último, la sobrenutrición conlleva elevación de ácidos grasos libres saturados e insaturados que también tienen características proinflamatorias activando células endoteliales vasculares, adipocitos o células mieloides.
También la inflamación, puede ocasionar, independientemente de la obesidad, resistencia a la acción de la insulina. Los mecanismos que han sido implicados en este efecto son:
- 1.
El TNFα ha sido relacionado con la RI en los estados inflamatorios. Sus efectos sobre el metabolismo lipídico, coagulación y función endotelial son determinantes en este sentido. Se sabe que puede tener efectos sobre el receptor de insulina, impidiendo el efecto de transducción de este tras ser estimulado por la insulina. También se conocen vías independientes del receptor de la insulina por los cuales el TNF induce RI, tales mecanismos son fundamentalmente la inhibición de genes como los de GLUT49.
- 2.
Del mismo modo, desde hace años, se conoce que el uso de determinados compuestos antiinflamatorios como los salicilatos, y concretamente la aspirina, tienen un efecto beneficioso sobre la hiperglucemia, efecto insulina sensibilizante. Esto se cree que ocurre porque tienen una acción interfiriendo sobre la vía del NF-kappa B, un factor de transcripción relacionado con inflamación.
- 3.
Otros factores como Jnk1 y 2 (C-jun-N-terminal kinase 1 y 2) parecen jugar también un papel clave en inflamación, obesidad y diabetes. Se conoce que sus niveles están elevados en ratones obesos con insulinorresistencia y que igualmente, ratones knock-out Jnk1−/−desarrollan menor insulinorresistencia tras una dieta rica en grasa10.
- 4.
El óxido nítrico, implicado en vasodilatación y otros procesos, juega un papel importante en RI. Se sabe que los estados proinflamatorios activan la expresión de Nos2, gen encargado de la síntesis de iNOS (inducible nitric oxide synthase), siendo este responsable, en última instancia, de la RI observada en estados de sepsis11.
- 5.
La IL-10 tiene un efecto antiinflamatorio inhibiendo la activación de NFkB inducida por TNF. En humanos se sabe que existe una clara relación entre niveles bajos de IL-10 y RI, lo cual ha llevado a pensar que esta interleuquina tiene efectos insulina sensibilizantes. Esto se ha comprobado parcialmente observando que ratones tratados con IL-10 no desarrollan RI cuando son tratados de forma simultánea con IL-612.
- 6.
Otro proteína que ha unido inflamación y RI es MCP1 (monocyte chemoattractant protein-1). MCP1 es una quimioquina sintetizada por macrófagos y endotelio que promueve el reclutamiento de monocitos en zonas dañadas o inflamadas. Se sabe que ratones knock-out para CCR2, receptor de MCP1, expresan menor infiltración macrofágica en el tejido adiposo, menor esteatosis hepática, mayor sensibilidad a la insulina y un menor aumento de peso tras dietas ricas en grasas. Este mismo efecto se ha visto con ratones tratados con un antagonista farmacológico de CCR213.
Así pues, la RI es un complejo estado metabólico con diferentes etiologías tales como obesidad, inflamación crónica… en el cual queda por definir la secuencia etiopatogénica que lleva a que la señal insulínica se procese defectuosamente. Cabría esperar que la elevada concentración de factores proinflamatorios como TNF, IL-6, MCP1, NO, etc. que ocurre en las enfermedades crónicas cause una incorrecta respuesta tisular a la insulina. Cuando la obesidad es el punto inicial se considera que la hipertrofia y muerte de adipocitos son los mecanismos principales de activación de la RI. Si no existe obesidad los mecanismos implicados en el tejido muscular y hepático parecen ser los determinantes a la hora del desarrollo de RI.
Resistencia insulínica y artritis reumatoideAunque en los pacientes con AR existe una elevada prevalencia de síndrome metabólico y de factores asociados como obesidad, dislipemia o alteración del metabolismo de la glucosa, pocos trabajos han estudiado específicamente la RI en esta enfermedad. En el estudio más amplio14, se reclutaron 94 pacientes con AR en los que se estudio la RI y la función de la célula beta mediante HOMA. Se concluyó que la RI (HOMA-IR) se relacionaba con parámetros de inflamación y actividad de la enfermedad como proteína C reactiva, velocidad de sedimentación globular y DAS28. Igualmente la RI guardaba relación con perímetro abdominal, la presencia de hipertensión arterial y el uso de diuréticos y beta-bloqueantes. Por otro lado, la función de la célula beta (HOMA-%B) mostró correlación inversa con DAS28, articulaciones inflamadas y dolorosas y correlacionó positivamente con el uso de esteroides. En este mismo estudio, los análisis de regresión múltiple mostraron que el factor más determinante con HOMA-IR fue la obesidad abdominal (39–56%), representando la actividad de la enfermedad un 5%. En cuanto al HOMA-%B, el factor más relacionado fue la dosis acumulada de esteroides (11–55%), mientras que la actividad de la enfermedad, determinada mediante DAS28 y articulaciones tumefactas y dolorosas, era responsable en un 19–25% para este deterioro de la función beta.
De forma similar, Chung et al15 estudiaron, también mediante test de HOMA, la RI en 104 pacientes con AR y lo compararon con 124 con lupus eritematoso sistémico. Demostraron que los pacientes con AR tienen un índice HOMA superior a los pacientes con lupus, que este se correlaciona con el peso ajustado por edad, sexo y uso de corticoides, y que este, del mismo modo, se correlaciona directamente con niveles de IL6, TNFα, proteína C reactiva, velocidad de sedimentación globular, y grado de calcificación coronaria.
La Montagna et al16 también encuentran una mayor presencia de RI en pacientes con AR, con una relación significativa entre RI y ateroesclerosis subclínica, debida fundamentalmente, según el análisis multivariante, a la dosis de esteroides.
Otros estudios17–19 también han demostrado una correlación positiva entre los niveles de proteína C reactiva y el índice HOMA en pacientes con AR, y le atribuye a este último un papel del 21% en la elevación de esta proteína de fase aguda; igualmente se ha demostrado una correlación clara entre RI, medida también mediante HOMA, con la edad, grosor de íntima media de carótida, HAQ score y las concentraciones de colesterol y triglicéridos. En lo que tiene que ver con variables analíticas, a parte de la correlación con proteína C reactiva, también se ha hallado una relación entre RI y valores de IL-220 en pacientes con AR.
El único estudio que contradice esta hipótesis de la relación RI y AR lo aporta García Díaz21 donde no se encontraron diferencias en los valores de HOMA y QUICKI entre pacientes con AR y controles (74 casos y controles). Tampoco hallaron relación de esta RI con la actividad de la enfermedad, la proteína C reactiva ni con la cuantificación del calcio coronario. Sólo se encontró relación entre RI y perímetro de cintura y enfermedad vascular subclínica mediante ecografía carotídea, características no directamente relacionadas con AR.
En lo concerniente al papel de diferentes terapias sobre la RI en pacientes con AR no existen estudios concluyentes. Dessein22 ha mostrado un efecto beneficioso del metotrexate sobre la resistencia y sensibilidad a la insulina mediante HOMA y QUICKI y ha considerado que este efecto viene dado por su acción antiinflamatoria, si bien concluye también que no se puede excluir que este efecto esté mediado parcialmente por otras acciones de los fármacos clásicos inductores de remisión sobre el perfil lipídico u otras áreas.
En lo que se refiere a tratamientos antiTNFα existen 10 estudios en la literatura sobre la influencia de estos sobre la RI que se resumen en la tabla 2. Todos ellos han usado métodos diferentes para determinar la RI y el número de pacientes (7 a 56) y la duración del tratamiento (de 120min a 1 año) ha sido muy variable… En 3 de ellos23–25 no se obtuvo mejoría en la RI con el tratamiento (dos de ellos usaron el método clamp considerado de referencia) mientras que en los restantes siete estudios26–32 sí hubo mejoría siempre medida mediantes métodos indirectos. Sólo en uno de ellos29, que utilizó el método clamp, obtuvo mejoría a las 14 semanas en la sensibilidad a la insulina. Datos propios no publicados tras estudiar a 16 pacientes con AR en tratamiento con anti-TNF no muestran relación entre el bloqueo de esta citoquina y la RI.
Estudios sobre la influencia de los tratamientos antiTNFα en la resistencia insulínica
| Referencia | n | AntiTNFα | Enfermedad | Seguimiento | Método | Resultado |
| Seriolo | 21 | Infliximab | Artritis reumaiode | 24 semanas | HOMA, QUICKI | Mejoría |
| Martinez-Abundis | 12 | Etanercept | Psoriasis | 2 semanas | Clamp | No mejoría |
| Oguz | 7 | Infliximab | Artritis reumaiode | 9 meses | HOMA | Mejoría |
| Rosevinge | 9 | Adalimumab | Artritis reumaiode | 8 semanas | Clamp | No mejoría |
| Lo | 56 | Etanercept | Síndrome metabólico | 4 semanas | HOMA | No mejoría |
| Huvers | 8 | Infliximab | Artritis reumaiode, espodilitis anquilosante, y enfermedad de Whipple | 6 semanas | Clamp | Mejoría |
| Tam | 19 | Infliximab | Artritis reumaiode | 14 semanas | HOMA | Mejoría |
| González-Gay | 27 | Infliximab | Artritis reumaiode | 120 minutos | HOMA, QUICKI | Mejoría |
| Seriolo | 38 | Etanercept e infliximab | Artritis reumaiode | 24 semanas | HOMA, QUICKI | Mejoría |
| Kiortsis | 27 | Infliximab | Artritis reumaiode | 24 semanas | HOMA, QUICKI | Mejoría |
| Ferraz-Amaro (en prensa) | 16 | Etanercept, infliximab y adalimumab | Artritis reumaiode | 1 año | HOMA | No mejoría |
HOMA, Homeostatic Model Assessment; QUICKI, Quanitative Insulin Sensitivity Check Index Clamp, Hyperinsulinemic Euglycemic Clamp
Los corticoides son responsables de intolerancia a la glucosa33 y, dado su amplio uso en la AR, cabría esperar que fueran responsables, al menos en parte, de la RI en estos enfermos. No obstante, varios estudios34–36 han encontrado una relación discreta o nula entre los corticoides y la presencia de síndrome metabólico o de RI en pacientes con AR. Estos datos sugieren que su acción beneficiosa sobre la inflamación contrarresta su efecto sobre la RI.
ConclusiónCada vez cobra más importancia el papel de la inflamación crónica sobre el desarrollo de arterioesclerosis y cada vez también parece más importante esta como determinante de mortalidad en pacientes con AR. A tenor de lo comentado parece amplia la evidencia que apoya una relación entre resistencia a la acción de la insulina, inflamación y AR tal y como ocurre en otras enfermedades crónicas. Por este motivo, dato que esta resistencia a la insulina juega un papel inicial en este daño vascular y parece ser, junto a otros mecanismos tales como disfunción endotelial, adhesión celuar, etc… un nexo de unión entre inflamación y arterioesclerosis, los reumatólogos debemos de conocerla, familiarizarnos con su concepto, saber medirla, relacionarla con otros parámetros de la enfermedad y considerarla como una manifestación sistémica más de nuestros pacientes con AR.
FinanciaciónFinanciado con FUNCIS 31/2007.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.


