

El síndrome de Schnitzler es un trastorno que se describió por primera vez en 1972. Se publicó en 1974 por Liliane Schnitzler, una dermatóloga francesa, como una entidad autónoma.
Se caracteriza por ser una enfermedad rara y adquirida, que comparte características comunes con un grupo de enfermedades hereditarias conocidas como síndromes autoinflamatorios. Se ha descrito con más frecuencia en varones entre la cuarta y la quinta décadas de la vida. Su etiopatogenia es desconocida, aunque se ha relacionado con situaciones como la alteración del equilibrio de la interleucina-1. Los síntomas que se presentan son fiebre, urticaria y artralgias; además, pueden hallarse adenopatías y un componente monoclonal IgM. Un 15-20% de los pacientes que presentan esta entidad evoluciona a un síndrome linfoproliferativo. Los tratamientos convencionales son ineficaces, incluyendo los antihistamínicos, los antiinflamatorios, los glucocorticoides y los inmunosupresores.
Presentamos el caso de un varón de 65 años sin antecedentes médicos, evaluado por presentar clínica de 10 años de evolución consistente en fiebre intermitente, dolores articulares diseminados de intensidad moderada y lesiones cutáneas pruriginosas tipo exantema maculopapular, localizadas en el tórax, el abdomen y las extremidades inferiores. Tres meses antes de nuestra evaluación desarrolló dolor lumbar irradiado a la extremidad inferior izquierda, de carácter no continuo, intenso, que llegó a impedir la deambulación. En la exploración general se evidenció la presencia de un exantema maculopapular en el tórax, el abdomen y las extremidades, así como signo de Lassegue positivo en el miembro inferior izquierdo.
Los exámenes de laboratorio, que incluían hemograma, bioquímica, coagulación, serología, marcadores tumorales, proteinograma e inmunoelectroforesis, fueron normales, excepto por la aparición de un pico monoclonal IgM kappa y un aumento de la velocidad de sedimentación globular (VSG).
La lesión cutánea fue biopsiada. El examen histológico mostró un infiltrado linfocitario perivascular superficial, dispersión intersticial, con algunos neutrófilos a lo largo de los haces de colágeno1. Se realizó una tomografía computarizada cérvico-toraco-abdominal, la cual no presentó alteraciones significativas.
Ante los diferentes hallazgos hematológicos, se realizó aspirado/biopsia de médula ósea, que fue compatible con enfermedad de Waldenström. El paciente se encuentra ahora en seguimiento en la consulta externa. No se le ha iniciado tratamiento quimioterápico y se ha mantenido solo con tratamiento sintomático.
El síndrome de Schnitzler es una entidad infrecuente de difícil diagnóstico, dada la ausencia de marcadores biológicos periféricos y la necesidad de la confirmación de dicho diagnóstico mediante una combinación de manifestaciones clínicas, analíticas y radiológicas, así como la exclusión de otras enfermedades.
Hasta la fecha, sobre la base de los criterios de Lipsker et al., no hay más de 60 casos descritos. Estos criterios incluyen presencia de una erupción urticarial cutánea y un componente IgM monoclonal, así como mínimo 2 de los siguientes síntomas o signos: fiebre, artralgias o artritis, dolor óseo, nódulos linfáticos palpables, esplenomegalia o hepatomegalia, VSG elevada y leucocitosis2,3.
Nuestro paciente cumple criterios de síndrome de Schnitzler, puesto que presenta características clínicas (erupción urticarial y fiebre) y alteraciones analíticas (pico monoclonal IgM y aumento de la VSG).
Entre las diversas manifestaciones descritas previamente como parte de la evolución de la enfermedad, otros autores incluyen el desarrollo de un síndrome linfoproliferativo como el linfoma linfoplasmocítico, el síndrome de Ritcher, así como el linfoma marginal4.
El diagnóstico diferencial de síndrome de Schnitzler se debe de hacer con diferentes entidades, siendo estas: crioglobulinemia, vasculitis urticarial hipocomplementémica, déficit adquirido del inhibidor de C1, síndrome hiper IgD y enfermedad de Still del adulto.
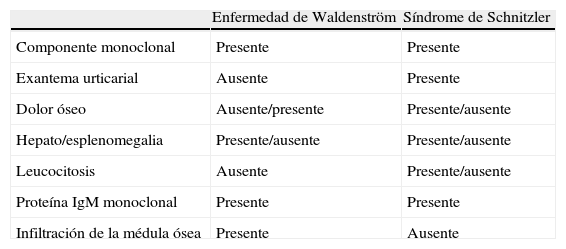
El interés de este artículo reside en que la presencia de un pico monoclonal IgM, junto con la histología de las lesiones cutáneas, fueron las alteraciones guía que condujeron al diagnóstico de síndrome de Schnitzler con evolución a enfermedad de Waldenström. Cabe hacer hincapié en que una buena anamnesis es fundamental, ya que permite conocer la naturaleza exacta de su evolución. Por último, destacamos la importancia de realizar un buen diagnóstico diferencial entre síndrome de Schnitzler1 y enfermedad de Waldeströn (tabla 1), así como la importancia de conocer esta entidad para evitar que el diagnóstico pase inadvertido.
Diferencias entre enfermedad de Waldeström y síndrome de Schintzler
| Enfermedad de Waldenström | Síndrome de Schnitzler | |
| Componente monoclonal | Presente | Presente |
| Exantema urticarial | Ausente | Presente |
| Dolor óseo | Ausente/presente | Presente/ausente |
| Hepato/esplenomegalia | Presente/ausente | Presente/ausente |
| Leucocitosis | Ausente | Presente/ausente |
| Proteína IgM monoclonal | Presente | Presente |
| Infiltración de la médula ósea | Presente | Ausente |


