

El síndrome de Hajdu-Cheney o síndrome acro-dento-osteo-displasia es una enfermedad rara caracterizada por osteólisis en banda de las falanges distales y dismorfia facial, entre otras manifestaciones. Describimos el caso de un varón de 45 años que consultó por dolor articular de características mecánicas en las manos, asociando dismorfia facial, alteraciones craneofaciales y deformidades digitales en telescopaje con acroosteólisis.
Hajdu-Cheney syndrome or acro-dento-osteo-dysplasia syndrome is a rare disease characterized by band osteolysis of distal phalanges and facial dysmorphia, among other manifestations. We present the case of a 45-year-old male who consulted for mechanical joint pain of both hands, facial dysmorphism, cranio-facial alterations, and digital telescoping with acroosteolysis.
El síndrome de Hajdu-Cheney (SHC) es una enfermedad rara inicialmente descrita por Hajdu en 1948 y más ampliamente estudiada por Cheney en 19651. La enfermedad se ha relacionado con mutaciones en el gen NOTCH2 localizado en el cromosoma 1 (1p13-p11)2.
La vía de señal NOTCH es muy relevante y altamente conservada en los organismos multicelulares, participando en múltiples procesos relacionados con el desarrollo y diferenciación tisular. Para su activación se precisa el contacto directo entre células adyacentes, habiéndose descubierto, hasta el momento, 4 tipos de receptores (NOTCH 1-4) y 5 ligandos (JAG1, JAG2, DLL1, DLL3 y DLL4). En el SHC se producen mutaciones con ganancia de función, determinando una acumulación de NOTCH2 que provoca señal excesiva sobre los osteoclastos, y que conduce a una resorción ósea marcada. Otro mecanismo adicional que contribuye a esta alteración es a través del incremento del TNFα y su acción promotora de diferenciación y activación osteoclástica. Por tanto, el SHC es una enfermedad que combina ambos procesos, tanto inflamación como destrucción ósea. En este sentido, al contrario que en el caso de SHC, la pérdida de función en el gen NOTCH se ha relacionado con una serie de enfermedades congénitas como el síndrome de Adams-Olivier o el síndrome de Alagille2,3.
En la mayoría de los casos la herencia es autosómica dominante, aunque se han descrito casos esporádicos2. Las manifestaciones clínicas más características afectan al macizo craneofacial y al esqueleto, aunque puede haber manifestaciones cardiológicas, neurológicas o renales4. Estos pacientes pueden acudir al reumatólogo por dolor articular mecánico, acroosteólisis y fracturas por fragilidad, dada su asociación con la osteoporosis5. En la actualidad no existe un tratamiento específico, siendo el abordaje habitual el manejo sintomático y de las complicaciones asociadas6. En este aspecto es habitual la indicación de antirresortivos como bisfosfonatos o denosumab para el tratamiento de la osteoporosis en estos pacientes. Recientemente se ha postulado el romosozumab como una alternativa atractiva, dado que es el fármaco que mayor respuesta densitométrica produce7.
Observación clínicaUn varón de 45 años, sin antecedentes personales ni familiares de interés, consultó por deformidad indolora de las manos y los pies, que en los últimos 2 años se acompañaba de dolor de características mecánicas. La anamnesis fue negativa para enfermedad reumática inflamatoria/autoinmune. El examen físico reveló pérdida dental completa, retrognatia, hipertelorismo y telescopaje digital en las manos y los pies. La analítica de sangre y orina, incluyendo reactantes de fase aguda y marcadores de autoinmunidad, fue normal. En el estudio radiológico presentaba osteólisis en banda en las falanges distales de las manos y los pies, asociando pseudofracturas de la 4.ª y 5.ª articulación metatarsofalángica (MTF) del pie izquierdo y la 5.ª MTF del pie derecho (fig. 1).
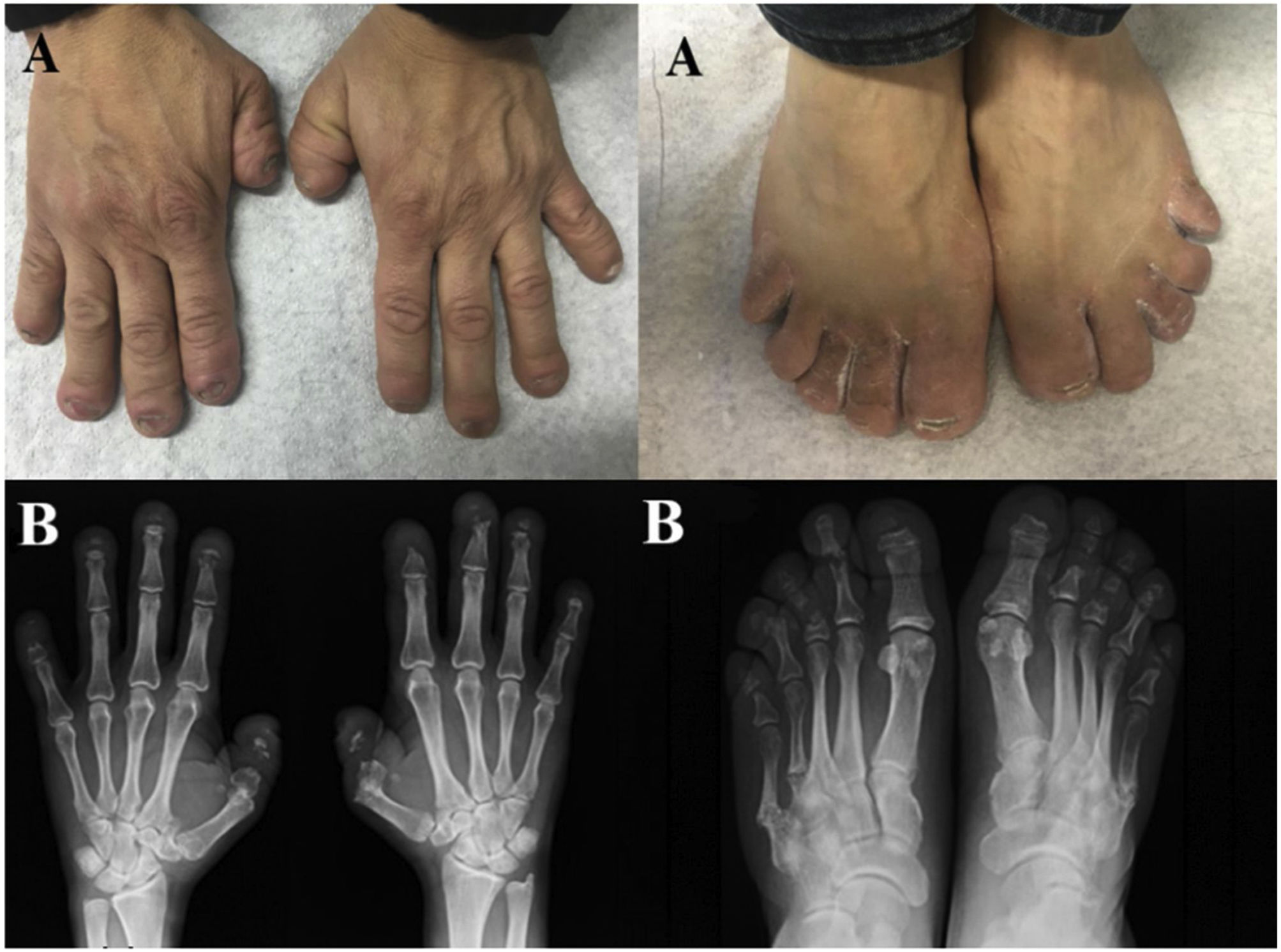
Ante estas pruebas de imagen y la ausencia de otros datos orientadores a enfermedades más convencionales, como artritis psoriásica (APs), se sospechó un síndrome de osteólisis congénito. El estudio genético detectó una mutación al nivel del exón 34 del gen NOTCH2 (variante p.Q2208X), diagnóstica de SHC. Los estudios de extensión detectaron una leve platibasia y osteoporosis vertebral densitométrica (Z-Score: 4DS). Actualmente el paciente recibe tratamiento analgésico y antirresortivo, sin precisar de intervención quirúrgica por la platibasia.
DiscusiónEl SHC constituye una rareza clínica (prevalencia estimada<1/1.000.000 habitantes) que sin embargo puede llegar a las consultas de reumatología y plantear un diagnóstico diferencial con entidades más comunes, y es por tanto necesario su conocimiento y manejo por parte del especialista1. La enfermedad obedece a mutaciones con ganancia de función localizadas en el exón 34 del gen NOTCH22. Aunque son múltiples las variantes estudiadas, la más común coincide con la objetivada en nuestro paciente (p.Q2208X)8. Esta variabilidad condiciona en los pacientes importantes diferencias a nivel fenotípico y de pronóstico, aunque en general todas ellas tienen en común la afectación dental, dismorfia facial y acroosteólisis1,5,7,8. Es habitual la asociación con osteoporosis por estimulación de las vías de señalización implicadas en el recambio óseo y la diferenciación de osteoclastos3,6,7,9,10. Las manifestaciones clínicas habitualmente comienzan en la infancia o en la pubertad, produciendo un deterioro funcional progresivo con el tiempo11. Es frecuente la afectación multisistémica, por lo que es recomendable realizar estudios de extensión de la enfermedad, como en nuestro caso, donde pudimos detectar una platibasia que podría eventualmente condicionar una invaginación basilar (complicación más frecuente y grave)5.
ConclusiónEl SHC es una entidad muy poco frecuente, donde es esencial una correcta anamnesis, exploración física y los test genéticos pertinentes. Los reumatólogos podemos colaborar no solo en el diagnóstico, sino también en el manejo de sus complicaciones, como la osteoporosis.
FinanciaciónNo se ha recibido financiación de otras entidades para la publicación de este artículo.
Conflicto de interesesLos autores declaran no presentar conflicto de intereses relacionados con este caso.


