





Se describe a una paciente de 51 años de edad con artritis reumatoide de 15 años de evolución, seropositiva –factor reumatoide positivo y anticuerpos antipéptido citrulinado positivos–, erosiva, no nodular, con poca adherencia al tratamiento y controles médicos, que presentó un cuadro caracterizado por pancitopenia persistente y hepatoesplenomegalia. La biopsia hepática y de médula ósea descartó tumores, amiloidosis e infecciones.
Se discute el diagnóstico diferencial de pancitopenia y hepatoesplenomegalia en una paciente con artritis reumatoide de larga evolución.
We describe the case of a 51-year-old woman with a seropositive, erosive, and non-nodular rheumatoid arthritis of 15 year of evolution. The patient had poor compliance with medical visits and treatment. She came to the clinic with persistent pancytopenia and spleen and liver enlargement. Liver and bone marrow biopsies were carried out and amyloidosis, neoplasias and infections were ruled out.
We discuss the differential diagnosis of pancytopenia and spleen and liver enlargement in a long-standing rheumatoid arthritis patient.
Se trata de una mujer de 51 años de edad con diagnóstico de artritis reumatoide (AR) que cumplía criterios del Colegio Americano de Reumatología de 1987, de 15 años de evolución, seropositiva (factor reumatoide [FR] y anticuerpos antipéptido citrulinado), erosiva, no nodular, que presentaba antecedentes familiares de linfoma. Negaba hábitos tóxicos y uso de drogas ilegales. Dicha paciente había realizado tratamiento con leflunomida (6 años), metotrexato (3 años) e hidroxicloroquina (un año) sin respuesta, por lo cual recibió 2 meses de tratamiento con etanercept, el cual se suspendió por reacción cutánea. Posteriormente recibió adalimumab durante 5 meses en el año 2010, perdiéndose del seguimiento.
En septiembre de 2012 consultó por astenia, pérdida de 8Kg de peso en 4 meses, disnea grado lll y distensión abdominal de 10 días de evolución. En el examen físico se presentó afebril (37°C), normotensa (tensión arterial 110/80mmHg), taquicárdica (110lpm), taquipneica (28 ciclos/min), adelgazada, con piel y mucosas pálidas con múltiples hematomas. El resto del examen mostró la presencia de un soplo sistólico 2/6 polifocal, hipoventilación y matidez pulmonar bibasal, abdomen globuloso, con hepatomegalia y esplenomegalia, ascitis y circulación colateral, así como una úlcera de 5cm de diámetro, dolorosa, en el trocánter de la cadera derecha, y lesiones purpúricas en los miembros inferiores, por debajo de las rodillas. En las manos se halló hipotrofia de músculos interóseos, desviación cubital, dedos en cuello de cisne y artritis en articulaciones metacarpofalángicas: el tercero de la derecha y el cuarto bilateral. La clinimetría mostró un HAQ: 2,375 y un DAS28: 7,29 (velocidad de sedimentación globular: 142mm en la primera hora, 3 articulaciones hinchadas, 12 articulaciones dolorosas, valoración actividad por paciente: 100mm).
Los datos de laboratorio mostraron: hematocrito 25%, hemoglobina 7,6g/dl, leucocitos 1.900 células/ml, neutrófilos 1.140 células/ml, plaquetas 82.000 células/ml, proteína C reactiva 17,4mg/l, velocidad de sedimentación globular 142mm en la primera hora, incremento de la fosfatasa alcalina (997U/l), FR (látex: 1/1.280), anticuerpos antipéptido citrulinado (53,7U/ml) y anticuerpos antinucleares (por Hep2): 1/2.560. La función renal, la alanina aminotransferasa (13U/l), la aspartato aminotransferasa (19U/l), los anticuerpos anticardiolipinas, el anticoagulante lúpico, los anti-ADN (Crithidia), los C3 y C4, y las serologías para histoplasmosis, VHB y VHC fueron normales.
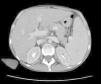
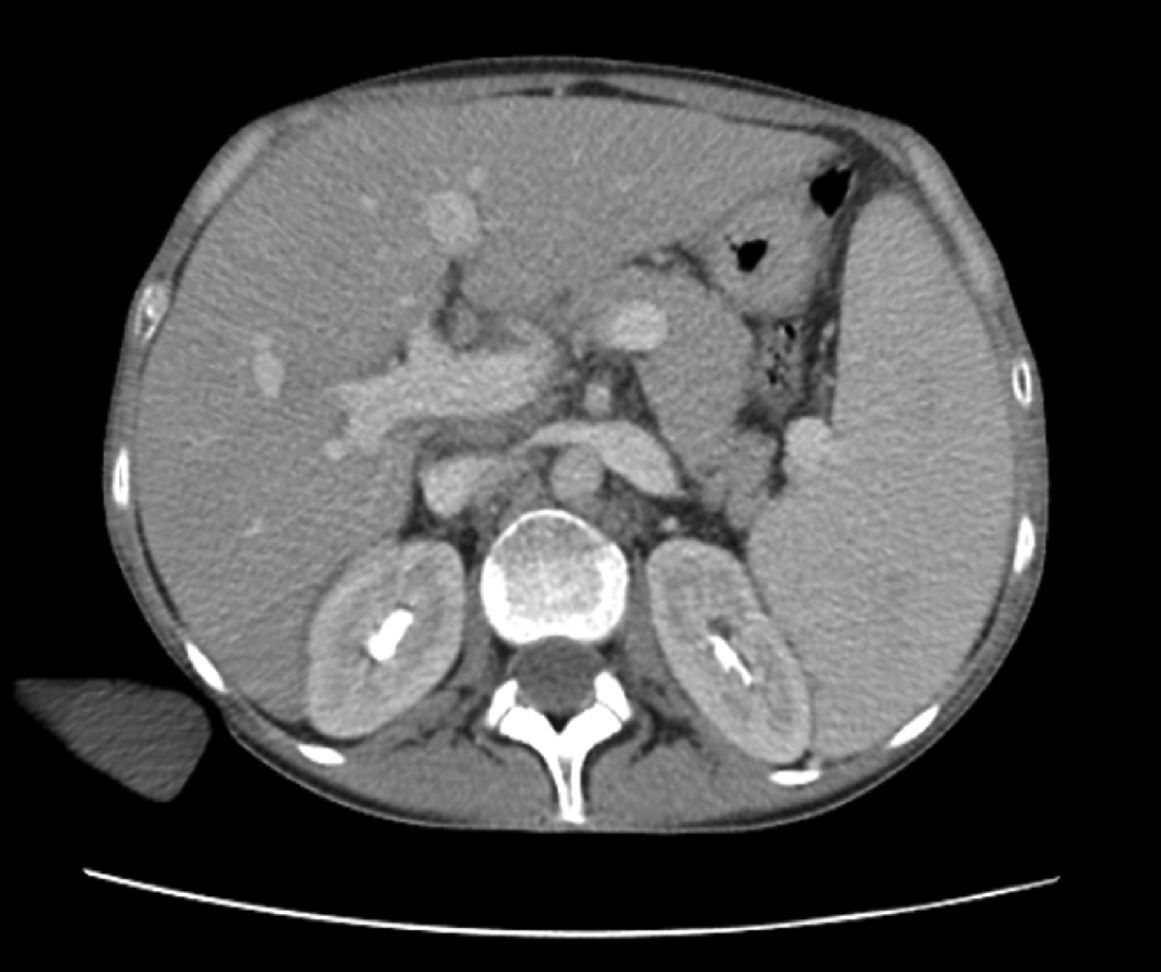
Los cultivos de sangre, orina, materia fecal y médula ósea fueron negativos para gérmenes comunes, micobacterias típicas y atípicas y hongos. La toracocentesis informó un exudado no complicado. Las tomografías computarizadas con contraste (oral e intravenoso) de cabeza y cuello, así como de pelvis, fueron normales; la de tórax mostró la presencia de un derrame pleural bibasal, y la de abdomen (fig. 1) mostró hepatomegalia homogénea (230mm de diámetro longitudinal) y esplenomegalia (200mm de diámetro longitudinal). El ecodoppler de vasos hepáticos mostró dilatación de la vena porta (14mm de diámetro) sin evidencias de trombosis. La fibroendoscopia digestiva alta descartó la presencia de varices esofágicas.
En el frotis de sangre periférica se observó leucopenia y trombocitopenia, por lo que se realizó una biopsia de médula ósea, donde se constató hipercelularidad con megacariocitos, hiperplasia de la serie mieloide y acúmulos aislados de linfocitos maduros. La paciente comenzó tratamiento con metilprednisolona 50mg/día por vía oral durante un mes, con posterior descenso gradual, metotrexato 15mg/semana, calcio, vitamina D y ácido fólico. Sin embargo, a pesar del tratamiento inmunosupresor, persistió la pancitopenia (hematocrito: 28,4%, hemoglobina: 9,2g/dl, leucocitos: 3.200 células/ml, plaquetas: 44.000 células/ml) y empeoró la función hepática (alanina aminotransferasa: 62U/l; aspartato aminotransferasa: 47U/l), por lo que se decidió realizar biopsia hepática.
Con base en los resultados obtenidos se decidió tratar a la paciente con meprednisona 10mg/día, metotrexato 10mg/semana, leflunomida 20mg/día, acido fólico, calcio y vitamina D, presentando la misma en la actualidad una AR inactiva con pancitopenia, pero sin neutropenia ni tendencia al sangrado.
Diagnóstico diferencialAnte una paciente con diagnóstico de AR de larga evolución que presenta pancitopenia persistente y hepatoesplenomegalia, el primer paso en el razonamiento diagnóstico que debería plantearse es si se debe a situaciones concomitantes, al tratamiento, o si es secundario a su enfermedad de base.
InfeccionesLos pacientes con AR tienen un riesgo aumentado de padecer infecciones1. Los principales factores de riesgo para su desarrollo son la presencia de manifestaciones extraarticulares, el padecimiento de comorbilidades, la edad avanzada, la leucopenia, y la terapia con corticoides y fármacos biológicos, entre otros2. Las infecciones más frecuentes son las de vías aéreas superiores, piel y partes blandas, huesos y articulaciones3.
La preocupación por el riesgo de infecciones severas y oportunistas (histoplasmosis, TBC, leishmaniasis, Pneumocystis carinii) entre los pacientes con enfermedades reumáticas se ha incrementado, sobre todo porque comparten varias características clínicas, tales como fiebre, fatiga, dolor de pecho, derrame pleural, infiltrados pulmonares difusos, pericarditis, mialgias, epistaxis, artralgias, artritis, eritema nudoso, pápulas difusas, lesiones en orofaringe, hepatoesplenomegalia, linfadenopatías, ACV, convulsiones, endocarditis, anemia, leucopenia, trombocitopenia, aumento de enzimas hepáticas y bilirrubina, y uveítis.
Ocasionalmente, la histoplasmosis se presenta primero con compromiso orgánico extrapulmonar. Estas lesiones aisladas se consideran usualmente manifestaciones de enfermedad diseminada, a pesar de la falta de compromiso pulmonar. En esta situación puede imitar a otras enfermedades, como el síndrome de Felty, y es importante sospecharla cuando ocurre una manifestación inusual en la enfermedad de base del paciente4,5.
Por lo tanto, si bien nuestra paciente procedía de zonas endémicas como el litoral argentino y la histoplasmosis puede simular un brote de la AR o una manifestación extraarticular de la misma (síndrome de Felty: astenia, artralgia, artritis, derrame pleural, hepatoesplenomegalia, pancitopenia y alteración del hepatograma), la serología, los hemocultivos y el cultivo de médula ósea e hígado fueron negativos para histoplasmosis y micosis profundas, lo que permitió descartar este diagnóstico.
NeoplasiasLa AR se caracteriza por una estimulación inmune persistente, la cual podría llevar a una proliferación linfocítica policlonal, con el aumento potencial de transformación maligna6. Según algunas comunicaciones, el riesgo de cáncer es 2 veces mayor en pacientes con AR comparado con el de la población general, siendo el riesgo estimado en estos pacientes para desarrollar linfoma de 1,5 a 8,77,mientras que el riesgo relativo de desarrollar linfoma no Hodgkin en el síndrome de Felty es cercano a 138. Los antagonistas del anti-TNF alfa no parecen aumentar la incidencia de linfoma9. La enfermedad actual de este caso engloba fundamentalmente una serie de síntomas hematológicos (pérdida de peso, hepatoesplenomegalia, leucopenia, anemia, trombocitopenia) que nos hicieron sospechar el viraje a linfoma. No obstante, la ausencia de adenopatías confirmada por tomografía computarizada y los resultados negativos en la biopsia de médula ósea y hepática nos permitieron excluirlos.
AmiloidosisOtra entidad rara de mal pronóstico asociada a AR de larga evolución que se puede presentar con síntomas sistémicos, hepatomegalia, miocardiopatía, neuropatía, lesiones purpúricas y proteinuria es la amiloidosis.
Se caracteriza por la acumulación extracelular de material amorfo, hialino y eosinofílico10. Su diagnóstico se establece mediante la tinción con Rojo Congo de mucosa rectal, grasa abdominal y órganos involucrados11. En esta paciente tal tinción no mostró la presencia de este material amorfo en tejido hepático.
Síndrome de FeltySe produce en<1% de las AR (de 10 a 15 años de evolución) con FR positivo, con enfermedad articular grave (erosiones, luxaciones) contrastando con inflamación articular moderada o ausente y acompañada de manifestaciones extraarticulares (pérdida de peso, pigmentación parda generalmente en la región pretibial, úlceras en los miembros inferiores, nódulos subcutáneos, linfadenopatías, síndrome de Sjögren y hepatoesplenomegalia).
El 60-70% se da en mujeres de 50 a 70 años de edad, y se caracteriza por la tríada de AR, neutropenia persistente (<2.000/mm3) sin otra causa que la justifique y esplenomegalia con una fuerte asociación con el haplotipo HLA-DR4 (casi el 95% de los casos)12–15.
Cabe considerar este diagnóstico como probable dado el tiempo de evolución (15 años) y las características de su enfermedad (FR positivo, erosiva), y acompañarse de neutropenia y esplenomegalia junto con pérdida de peso, lesiones pigmentadas y úlceras en miembros inferiores.
Síndrome de seudo-FeltyLa proliferación de linfocitos grandes granulares, también llamada síndrome de seudo-Felty, es una complicación sistémica rara (<0,6%) de la AR. Se caracteriza por la presencia de neutropenia persistente, linfocitosis y esplenomegalia, que de no mediar tratamiento adecuado puede progresar en un 3 a 14% de los casos a leucemia de linfocitos grandes granulares.
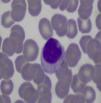
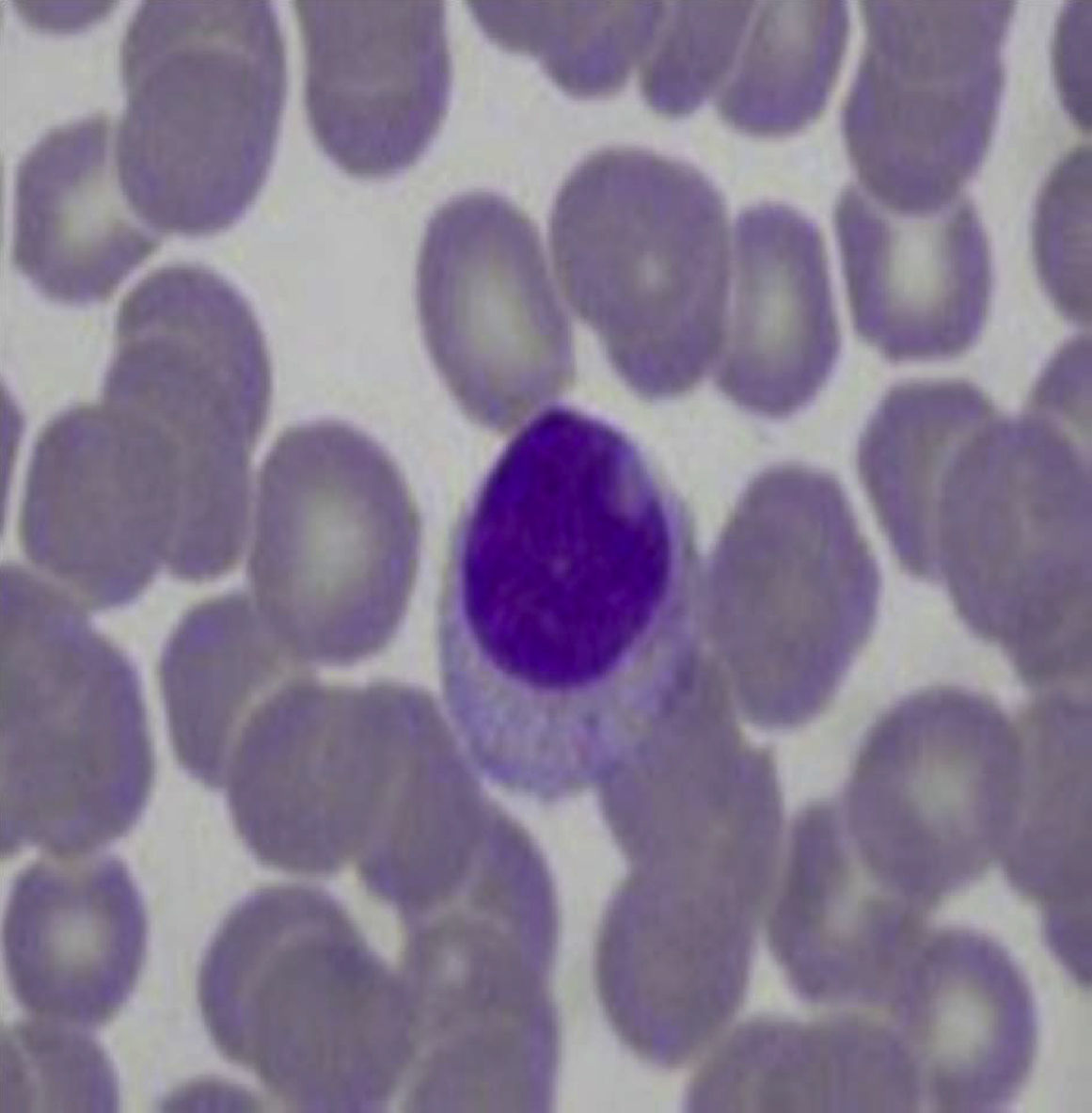
Aproximadamente el 30-40% de los pacientes con síndrome de Felty tiene expansiones de linfocitos grandes granulares en sangre periférica (linfocitosis>0,5×109/l) (tabla 1)12,13,16,17. Los mismos representan del 5 al 10% de las células mononucleares circulantes, y morfológicamente son de gran tamaño (15-18μ de diámetro), núcleo redondo o dentado, y citoplasma abundante con gránulos azurófilos (fig. 2)18. Cuando se asocia a invasión clonal de médula ósea, bazo o hígado, el cuadro recibe el nombre de leucemia de linfocitos grandes granulares. Esta es una enfermedad maligna de bajo grado, que se acompaña de neutropenia, anemia, trombocitopenia y alta susceptibilidad a infecciones19. La paciente de este caso presentaba una AR de larga evolución con daño articular severo y secuelas importantes, acompañada de manifestaciones clínicas similares al síndrome de Felty, pero sin linfocitosis ni linfocitos grandes granulares en el frotis de sangre periférica, por lo que se descartó este diagnóstico.
Diferencias clínicas y de laboratorio entre el síndrome Felty y el seudo-Felty
| Síndrome de Felty | Síndrome de seudo-Felty | |
|---|---|---|
| Manifestaciones extraarticulares | Común | Común |
| Artritis erosiva | Común | Común |
| Infecciones recurrentes | Común | Común |
| Esplenomegalia | Común | Común |
| Progresión a leucemia | Raro | 3-14% |
| Remisión espontánea | 0-22% | 0-14% |
| Recuento de leucocitos | Bajo | Normal/bajo |
| Linfocitosis | Ausente | Presente |
| FR, ANA (positivos) | Común | Común |
| Respuesta a la esplenectomía | Mejora | Exacerbación |
La presencia de enfermedad hepática significativa en pacientes con AR no es frecuente. Cuando se presenta una hepatopatía significativa, generalmente se debe a un compromiso sistémico autoinmune que también compromete el hígado, o a coinfección con virus hepatotropos como el B y el C, o hepatotoxicidad por el tratamiento20.
La hiperplasia nodular regenerativa hepática (HNRH) es un trastorno raro descrito por primera vez por Ranstrom en 1953 como «adenomatosis miliar hepatocelular», y tiene muchos sinónimos, incluyendo la hipertensión portal no cirrótica, la hiperplasia nodular difusa y la transformación nodular del hígado21. Steiner Coind la denominó hiperplasia nodular regenerativa, término actualmente aceptado para esta lesión caracterizada por nódulos hepáticos secundarios a hiperplasia hepatocitaria con ausencia o escasa fibrosis e hipertensión portal. El pronóstico generalmente es bueno, a diferencia de la hipertensión portal debida a cirrosis. Ambas pueden confundirse fácilmente, por ello es fundamental la biopsia hepática22.
Diagnóstico clínico del presentadorNuestra paciente tenía las características clínicas de una AR asociada a síndrome de Felty.
Integrando estos datos con el ecodoppler de vasos hepáticos, que mostraba dilatación de la vena porta con signos de hipertensión portal (ascitis y pancitopenia periférica secundaria a esplenomegalia), y habiendo sido descartadas otras causas de hepatoesplenomegalia, asumo que la paciente presenta una HNRH como afección autoinmunitaria secundaria a su enfermedad de base, siendo la biopsia hepática la prueba diagnóstica necesaria para confirmar su diagnóstico.
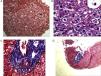
Resultado definitivo y comentarioLa biopsia hepática constituyó una prueba diagnóstica, demostrándose la presencia de nodularidad del parénquima y la regeneración hepatocitaria, con espacios porta conservados, sin evidencia de fibrosis significativa y tinción con Rojo Congo negativa (fig. 3).
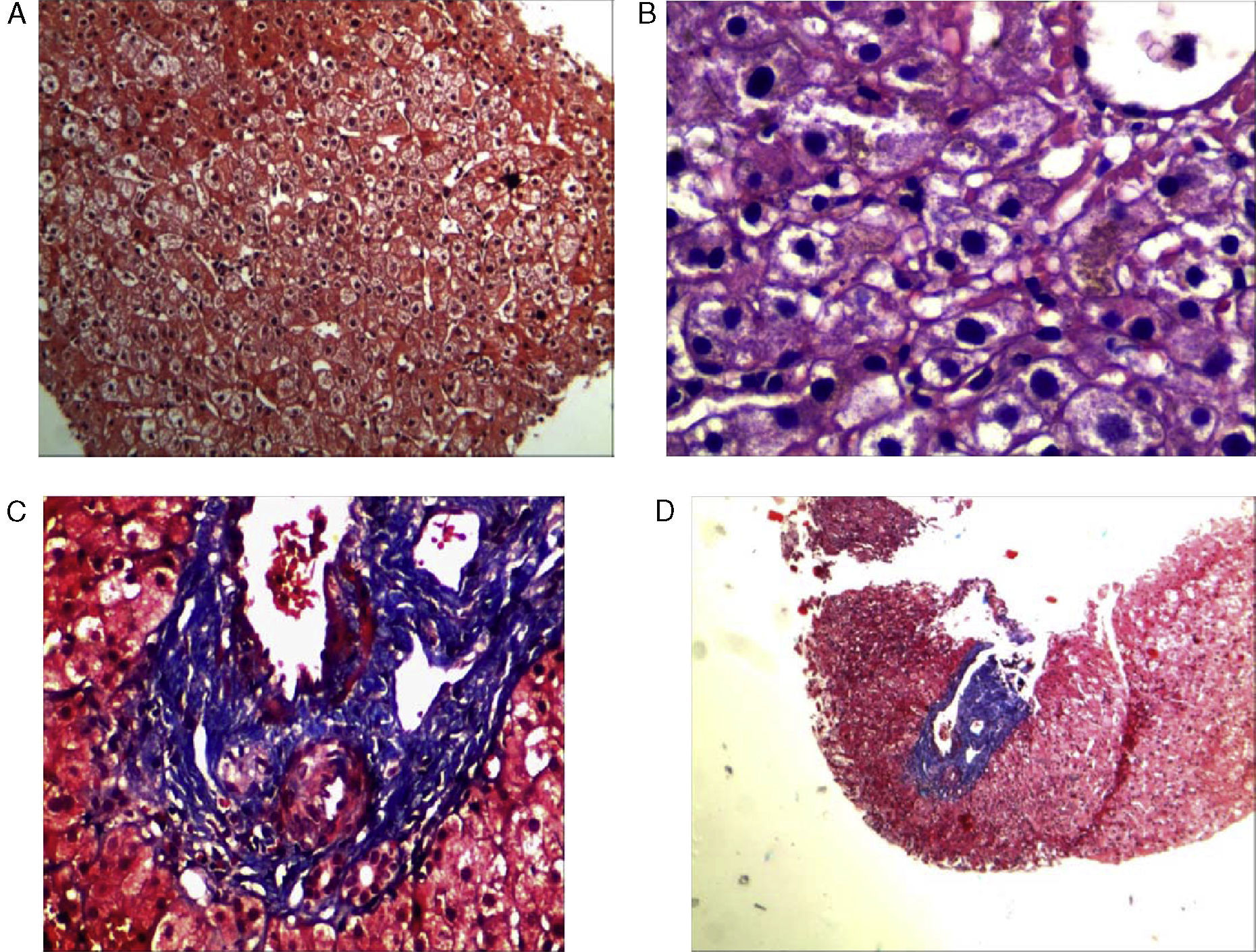
La hiperplasia nodular regenerativa es una entidad poco frecuente que afecta por igual a ambos sexos y generalmente se encuentra en asociación con múltiples enfermedades (tabla 2), siendo la AR y el síndrome de Felty las más frecuentes21,23,24. Los pacientes con HNRH pueden estar asintomáticos o presentarse con dolor abdominal recurrente, síntomas sistémicos, signos de hiperesplenismo (esplenomegalia o anormalidades hematológicas) y signos de hipertensión portal (tales como ascitis, varices esofágicas sangrantes o esplenomegalia). Las pruebas de laboratorio presentan alteración leve del hepatograma de una manera no específica (elevación de aspartato aminotransferasa, alanina aminotransferasa, GGT y principalmente fosfatasa alcalina).
Condiciones asociadas con hiperplasia nodular regenerativa hepática
| Reumatológicas | Hematológicas | Fármacológicas | Congénitas | Otras |
|---|---|---|---|---|
| AR | PTI | Azatioprina | Agenesia de vena porta | Síndrome del aceite tóxico |
| Síndrome de Felty | Policitemia vera | Busulfán | Anomalías cardiacas | Metástasis |
| LES | Trombocitosis esencial | Doxorrubicina | CBP | |
| PAN | Anemia de células falciformes | Ciclofosfamida | Enfermedad celíaca | |
| Esclerosis sistémica | Macroglobulinemia | Clorambucilo | ICC | |
| SAAF | Metaplasia mieloide | Bleomicina | TBC | |
| Síndrome CREST | Leucemia linfocítica | |||
| Síndrome POEMS | Linfoma Hodgkin y no Hodgkin |
AR: artritis reumatoide; CBP: cirrosis biliar primaria; ICC: insuficiencia cardiaca congestiva; LES: lupus eritematoso sistémico; PAN: panarteritis nudosa; POEMS: polineuropatía+organomegalia+proteína M+organomegalia+alteraciones cutáneas; PTI: púrpura trombocitopénica idiopática; SAAF: síndrome antifosfolipídico; TBC: tuberculosis.
Tomada de Malnick et al.23.
Un 68% de los pacientes presentan anticuerpos antinucleares positivos y un porcentaje similar presenta positividad para el FR. Con respecto a la patogenia, se han propuestos varias hipótesis: a) incremento del flujo portal debido a un aumento del flujo esplénico; b) daño vascular primario, por ejemplo, vasculitis asociada a AR, y c) flujo hepático alterado debido a la isquemia hepática.
En 1998, Pérez Ruiz et al. mostraron un posible papel de los anticuerpos antifosfolipídicos en la patogenia24.
Por otro lado, hallazgos normales en la biopsia con aguja no excluyen el diagnóstico, ya que si bien la HNRH compromete difusamente el hígado, lo hace de manera parcheada y el grado de nodularidad puede variar de una porción del hígado a otra. El tratamiento se centra en corregir la causa desencadenante (enfermedad autoinmune, trastornos hematológicos, fármacos).
Si bien estos pacientes presentan un pronóstico relativamente benigno en comparación con la hipertensión portal de causas cirróticas, una serie de pacientes han requerido esplenectomía y derivación portal para resolver complicaciones tales como pancitopenia y hemorragias digestivas secundarias a varices esofágicas. El trasplante hepático es rara vez necesario y se reserva para pacientes con insuficiencia hepática25,26.
En conclusión, la HNRH es una complicación que puede aparecer durante la evolución de las enfermedades autoinmunes, por lo que debería considerarse en los casos que presentan hepatomegalia, alteraciones persistentes del hepatograma y/o signos de hipertensión portal.
Responsabilidades éticasProtección de personas y animalesLos autores declaran que para esta investigación no se han realizado experimentos en seres humanos ni en animales.
Confidencialidad de los datosLos autores declaran que en este artículo no aparecen datos de pacientes.
Derecho a la privacidad y consentimiento informadoLos autores han obtenido el consentimiento informado de los pacientes y/o sujetos referidos en el artículo. Este documento obra en poder del autor de correspondencia.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.


