


El uso de biológicos ha permitido conocer de manera exhaustiva su seguridad gracias a registros como BIOBADASER. El presente trabajo permite, con un estudio observacional de cohortes, describir el perfil de seguridad perinfusional de dichos tratamientos por vía intravenosa.
ObjetivosConocer el perfil de seguridad en la práctica clínica, tras la administración de biológicos por vía intravenosa y durante las 24 h posteriores.
Material y métodosCohorte transversal de 114 pacientes con AR tratados con agentes biológicos (criterios ACR) durante un mes de 2009 por enfermería de hospital de día de 12 centros hospitalarios catalanes. Se analizaron la edad, el sexo, los tratamientos actuales y previos, los datos de vacunación previa y la premedicación. Se registró también cualquier acontecimiento adverso (AA) durante la administración o en las 24 h posteriores. Se clasificó según el diccionario internacional MedDRAv11.0 y se describieron la intensidad (leve, moderada, severa), la relación con la administración del fármaco según el algoritmo de Karch y Lasagna (no relacionada, improbable, posible, probable, definitiva) y las medidas emprendidas. El análisis estadístico se realizó mediante SPSS 18.0.
ResultadosCiento once con criterios de inclusión (edad media ± desviación estándar 56,06 ± 12,12 años), 90 mujeres (81,1%) y evolución de 11,97 ± 7,95 años; 24 pacientes (21,6%) con antecedentes de alergia. Se observaron 12 AA en 7 pacientes, 9 de ellos durante la administración y 3 en las 24 h posteriores. No hubo ningún acontecimiento adverso grave y uno de los AA se calificó de intensidad moderada (urticaria). El resto de los AA fueron de intensidad leve.
The Biologics used in the management of rheumatoid arthritis (RA) in recent years, have comprehensively permitted to understand its security, as shown in registries such as BIOBADASER. The present manuscript represents an observational cohort study to describe the safety perinfusional profile of those intravenous treatments.
ObjectivesTo confirm the safety profile of biological therapies in routine clinical practice, after the administration of intravenous drugs and 24hours post-administration.
Material and methodsWe evaluated a cross-sectional cohort of 114 patients with RA (according to the American College of Rheumatology ACR criteria), attending within one month in 2009 the nursing clinics of day care hospital of 12 Catalonian hospitals. All patients were treated with intravenous biological agents. We recorded the age, sex, current and previous drug treatments, we also collected data about previous vaccination and premedication received and any adverse event occurring at the time of drug administration or within 24hours. If an adverse event occurred, was categorized by MedDRAv11.0 International Dictionary, and categorized in terms of intensity (mild, moderate, severe), relationship to drug administration according to Karch and Lasagna algorithm (unrelated, unlikely, possible, probable, definite) and the further measures taken.
Results111 patients met the inclusion criteria, with a mean age of 56.06 years (SD: 12.12), 90 of them women (81.1%) and mean time since diagnosis of the disease of 11.97 years (SD: 7.95). 24 patients (21.6%) had a history of allergy. 12 adverse events were observed in 7 patients, 9 of which at the time of administration and 3 in 24hours after. There were no serious adverse events and only one of the adverse events (AEs) was rated as moderate (urticaria). The remaining AA were mild.
La artritis reumatoide (AR) es una enfermedad inflamatoria crónica, que cursa con destrucción progresiva y presenta inflamación, dolor, rigidez matutina y pérdida de capacidad funcional, afectando sobre todo a manos y pies. La prevalencia en nuestro país es del 0,5%, lo que representa unos 200.000 adultos mayores de 20 años afectados1,2. La pérdida funcional de las articulaciones disminuye la capacidad para la realización de las actividades de la vida diaria, provoca problemas en el ámbito laboral y social, y limita considerablemente la calidad de vida relacionada con la salud. La mortalidad relacionada con la AR es superior a la de la población general y supone una disminución en la esperanza de vida entre 5 y 10 años3,4. Los costes derivados de la enfermedad suponen un enorme consumo de recursos sanitarios y sociales, estimado anualmente de 1.120 millones de euros para estos 200.000 pacientes4,5.
El objetivo actual del tratamiento de la AR es inducir la remisión o, en su defecto, alcanzar una enfermedad de baja actividad inflamatoria. El tratamiento precoz y activo en las fases más iniciales de la enfermedad es fundamental para obtener buen pronóstico a largo plazo5,6. Entre los tratamientos existentes, las terapias biológicas han representado un enorme avance en el tratamiento de la AR.
La función de enfermería en el hospital de día de Reumatología es administrar y controlar la terapia biológica, detectar precozmente los efectos secundarios, monitorizar la comorbilidad e informar al paciente sobre los signos de alarma que debe vigilar en relación al tratamiento, así como la instrucción al paciente de la autoadministración de fármacos biológicos subcutáneos7,8. Los fármacos biológicos por vía intravenosa más utilizados en el hospital de día son infliximab (Remicade®), rituximab (Mabthera®), abatacept (Orencia®) y tocilizumab (Roactemra®).
El presente trabajo pretende evaluar el perfil de seguridad de las terapias biológicas en condiciones de práctica clínica habitual, tras la administración de fármacos por vía intravenosa y durante las 24 h posteriores a la administración del mismo, y describir el papel de enfermería en todo el proceso de la enfermedad y su tratamiento.
Material y métodoDe los 114 pacientes reclutados, se analizó una cohorte transversal de 111 pacientes, con una edad media ± desviación estándar de 56,06 ± 12,1 años, 90 (81,1%) mujeres, 21 (18,9%) hombres, y una evolución de la enfermedad desde el momento del diagnóstico de 11.97 ± 7,95 años. Ciento once cumplían criterios de AR según el American College of Rheumatology (ACR), y fueron evaluados durante el periodo comprendido entre el 15 de noviembre y el 15 de diciembre de 2009 en hospitales de día y consultas de enfermería de 12 centros hospitalarios de Catalunya. Todos los pacientes firmaron un consentimiento informado por escrito para participar en el estudio. Se registraron en un cuaderno las variables demográficas: edad y sexo de los pacientes, los tratamientos farmacológicos actuales y previos, así como los datos de la vacunación previa y premedicación (tablas 1-2). Se registró cualquier acontecimiento adverso (AA) ocurrido en el momento de la administración del fármaco o en las 24 h posteriores. Los AA registrados se clasificaron según el diccionario internacional MedDRAv11.0, describiendo el grado de intensidad (leve, moderada, severa); se valoraron la relación con la administración del fármaco según el algoritmo de Karch y Lasagna (no relacionada, improbable, posible, probable, definitiva) y las medidas tomadas. Los procedimientos realizados antes, durante y después de la administración del fármaco por parte de enfermería se registraron para monitorizar el proceso. Se realizaron una valoración y una monitorización hemodinámica del paciente; se canalizó una vía periférica, por la que tras la extracción analítica de control, se inició la administración del tratamiento por vía intravenosa con bomba de perfusión continua según protocolo. La administración del fármaco se realizó mediante bomba de perfusión por vía intravenosa en 88 pacientes (80-0%) y mediante gravedad en 22 (20%) pacientes, con una media de infusiones de 18,3 y una mediana de 9,5.
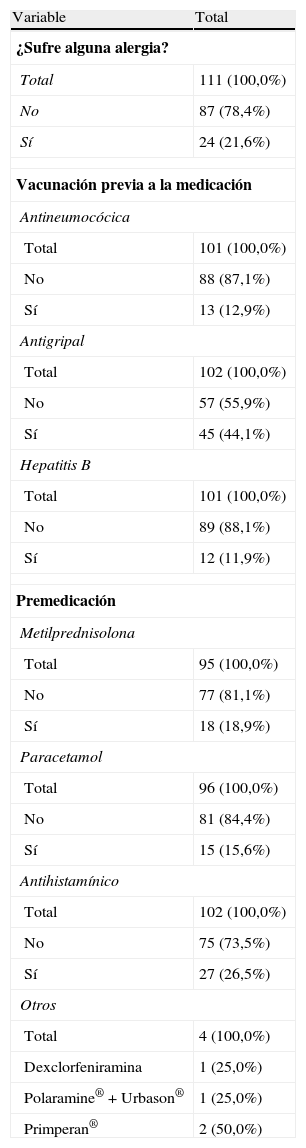
Parámetros clínicos: alergias, vacunación y premedicación
| Variable | Total |
| ¿Sufre alguna alergia? | |
| Total | 111 (100,0%) |
| No | 87 (78,4%) |
| Sí | 24 (21,6%) |
| Vacunación previa a la medicación | |
| Antineumocócica | |
| Total | 101 (100,0%) |
| No | 88 (87,1%) |
| Sí | 13 (12,9%) |
| Antigripal | |
| Total | 102 (100,0%) |
| No | 57 (55,9%) |
| Sí | 45 (44,1%) |
| Hepatitis B | |
| Total | 101 (100,0%) |
| No | 89 (88,1%) |
| Sí | 12 (11,9%) |
| Premedicación | |
| Metilprednisolona | |
| Total | 95 (100,0%) |
| No | 77 (81,1%) |
| Sí | 18 (18,9%) |
| Paracetamol | |
| Total | 96 (100,0%) |
| No | 81 (84,4%) |
| Sí | 15 (15,6%) |
| Antihistamínico | |
| Total | 102 (100,0%) |
| No | 75 (73,5%) |
| Sí | 27 (26,5%) |
| Otros | |
| Total | 4 (100,0%) |
| Dexclorfeniramina | 1 (25,0%) |
| Polaramine®+Urbason® | 1 (25,0%) |
| Primperan® | 2 (50,0%) |
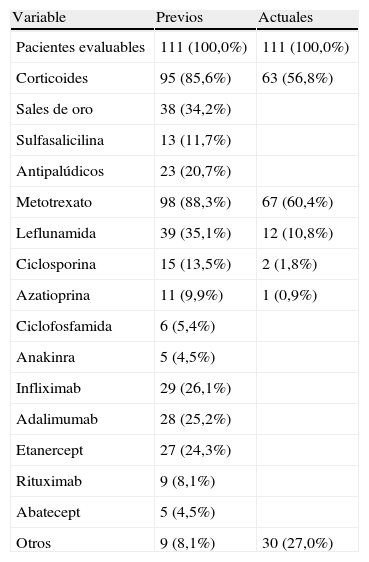
Tratamientos previos y tratamientos actuales de la AR
| Variable | Previos | Actuales |
| Pacientes evaluables | 111 (100,0%) | 111 (100,0%) |
| Corticoides | 95 (85,6%) | 63 (56,8%) |
| Sales de oro | 38 (34,2%) | |
| Sulfasalicilina | 13 (11,7%) | |
| Antipalúdicos | 23 (20,7%) | |
| Metotrexato | 98 (88,3%) | 67 (60,4%) |
| Leflunamida | 39 (35,1%) | 12 (10,8%) |
| Ciclosporina | 15 (13,5%) | 2 (1,8%) |
| Azatioprina | 11 (9,9%) | 1 (0,9%) |
| Ciclofosfamida | 6 (5,4%) | |
| Anakinra | 5 (4,5%) | |
| Infliximab | 29 (26,1%) | |
| Adalimumab | 28 (25,2%) | |
| Etanercept | 27 (24,3%) | |
| Rituximab | 9 (8,1%) | |
| Abatecept | 5 (4,5%) | |
| Otros | 9 (8,1%) | 30 (27,0%) |
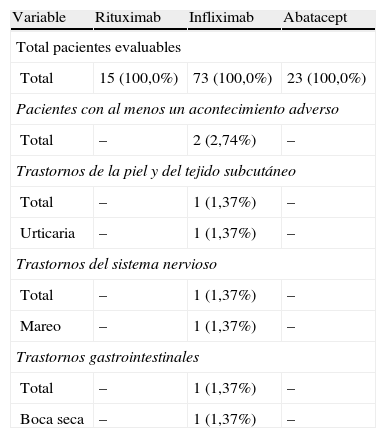
Durante la administración o tras 24 h, de los 111 pacientes que cumplieron los criterios de inclusión, 24 pacientes (21,6%) presentaban antecedentes de alergia. Se observaron un total de 12 AA en 7 pacientes, 9 de los cuales en el momento de la administración y 3 en las 24 h posteriores (tabla 3). No se observó ningún AA grave y solo uno de los AA se calificó de intensidad moderada (urticaria). Los AA restantes fueron de intensidad leve. Las características de la reacción durante y tras la infusión, y los fármacos implicados se presentan en las tablas 4–7.
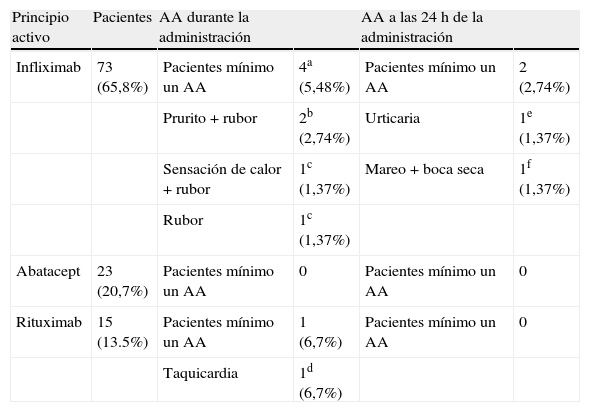
Acontecimientos adversos durante la administración y a las 24 horas: relación con el fármaco y medidas adoptadas
| Principio activo | Pacientes | AA durante la administración | AA a las 24 h de la administración | ||
| Infliximab | 73 (65,8%) | Pacientes mínimo un AA | 4a (5,48%) | Pacientes mínimo un AA | 2 (2,74%) |
| Prurito+rubor | 2b (2,74%) | Urticaria | 1e (1,37%) | ||
| Sensación de calor+rubor | 1c (1,37%) | Mareo+boca seca | 1f (1,37%) | ||
| Rubor | 1c (1,37%) | ||||
| Abatacept | 23 (20,7%) | Pacientes mínimo un AA | 0 | Pacientes mínimo un AA | 0 |
| Rituximab | 15 (13.5%) | Pacientes mínimo un AA | 1 (6,7%) | Pacientes mínimo un AA | 0 |
| Taquicardia | 1d (6,7%) |
a Todos presentaron, al menos, rubor facial.
bUn caso con relación probable con la administración del fármaco (se interrumpió temporalmente el tratamiento) y el otro con relación definitiva (se ajustó la dosis).
cRelación definitiva con la medicación (se interrumpió temporalmente el tratamiento).
dRelación posible con la medicación (ninguna acción emprendida).
eRelación probable con la medicación (ninguna acción emprendida).
fRelación definitiva con la medicación (ninguna acción emprendida).
El manejo de los acontecimientos adversos (AA) se realizo mediante la monitorización clínica y hemodinámica exhaustiva de los pacientes, hasta la resolución del cuadro agudo. No se retiró ningún tratamiento por AA grave.
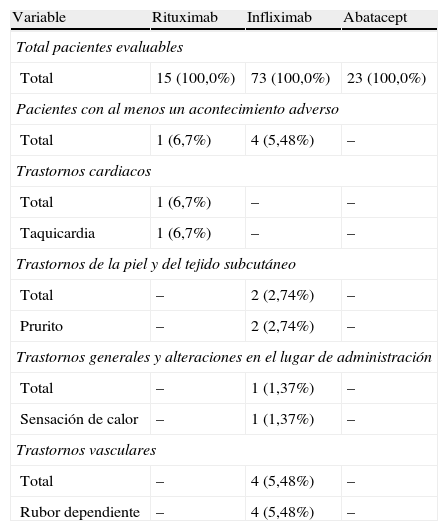
Acontecimientos adversos durante la administración del fármaco por tratamiento actual
| Variable | Rituximab | Infliximab | Abatacept |
| Total pacientes evaluables | |||
| Total | 15 (100,0%) | 73 (100,0%) | 23 (100,0%) |
| Pacientes con al menos un acontecimiento adverso | |||
| Total | 1 (6,7%) | 4 (5,48%) | – |
| Trastornos cardiacos | |||
| Total | 1 (6,7%) | – | – |
| Taquicardia | 1 (6,7%) | – | – |
| Trastornos de la piel y del tejido subcutáneo | |||
| Total | – | 2 (2,74%) | – |
| Prurito | – | 2 (2,74%) | – |
| Trastornos generales y alteraciones en el lugar de administración | |||
| Total | – | 1 (1,37%) | – |
| Sensación de calor | – | 1 (1,37%) | – |
| Trastornos vasculares | |||
| Total | – | 4 (5,48%) | – |
| Rubor dependiente | – | 4 (5,48%) | – |
Características de los acontecimientos adversos durante la administración del fármaco
| Variable | Total |
| Intensidad | |
| Total | 5 (100,0%) |
| Leve | 5 (100,0%) |
| Valoración de la causalidad del AA con la administración del fármaco | |
| Total | 5 (100,0%) |
| Definitiva | 3 (60,0%) |
| Posible | 1 (20,0%) |
| Probable | 1 (20,0%) |
| Medidas que ha obligado a emprender | |
| Total | 5 (100,0%) |
| Ajuste de la dosis de la medicación del estudio | 1 (20,0%) |
| Interrupción temporal de la medicación del estudio | 3 (60,0%) |
| Ninguna acción | 1 (20,0%) |
| ¿Es un AA grave? | |
| Total | 4 (100,0%) |
| No | 4 (100,0%) |
Acontecimientos adversos tras 24 h de la administración del fármaco por tratamiento
| Variable | Rituximab | Infliximab | Abatacept |
| Total pacientes evaluables | |||
| Total | 15 (100,0%) | 73 (100,0%) | 23 (100,0%) |
| Pacientes con al menos un acontecimiento adverso | |||
| Total | – | 2 (2,74%) | – |
| Trastornos de la piel y del tejido subcutáneo | |||
| Total | – | 1 (1,37%) | – |
| Urticaria | – | 1 (1,37%) | – |
| Trastornos del sistema nervioso | |||
| Total | – | 1 (1,37%) | – |
| Mareo | – | 1 (1,37%) | – |
| Trastornos gastrointestinales | |||
| Total | – | 1 (1,37%) | – |
| Boca seca | – | 1 (1,37%) | – |
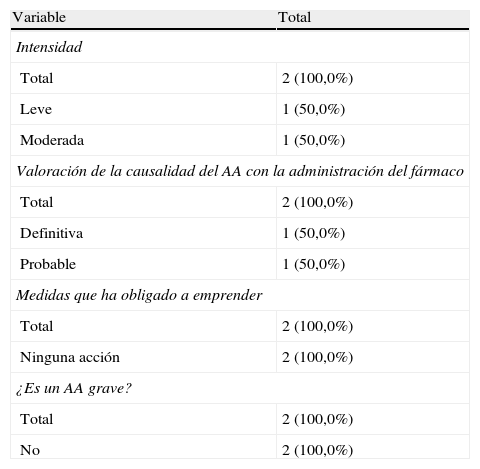
Características de los acontecimientos adversos tras 24 h de la administración del fármaco
| Variable | Total |
| Intensidad | |
| Total | 2 (100,0%) |
| Leve | 1 (50,0%) |
| Moderada | 1 (50,0%) |
| Valoración de la causalidad del AA con la administración del fármaco | |
| Total | 2 (100,0%) |
| Definitiva | 1 (50,0%) |
| Probable | 1 (50,0%) |
| Medidas que ha obligado a emprender | |
| Total | 2 (100,0%) |
| Ninguna acción | 2 (100,0%) |
| ¿Es un AA grave? | |
| Total | 2 (100,0%) |
| No | 2 (100,0%) |
Las terapias biológicas por vía intravenosa con infliximab, rituximab, abatacept y tocilizumab son eficaces en el tratamiento de la AR y otras enfermedades inflamatorias crónicas que cursan con brotes de reagudización. Infliximab, un anti-TNF-α, de administración por vía intravenosa con una frecuencia de cada 8 semanas, es a día de hoy el más utilizado y con mayor experiencia en hospital de día. Rituximab es un anticuerpo monoclonal quimérico que actúa sobre los linfocitos B; su administración se realiza en 2 ciclos anuales, cada 6 meses aproximadamente, y su manejo protocolizado en hospital de día requiere de la administración de premedicación. Menos frecuentemente, abatacept que es una proteína obtenida por cultivo celular que bloquea la coestimulación de linfocitos T; se administra también por vía intravenosa mensualmente. Entre las complicaciones que se presentan con mayor frecuencia en este tipo de terapias se encuentran los AA relacionados con su administración. Estos AA pueden clasificarse en reacciones agudas o tardías, según se presenten en el momento de la administración o en las siguientes 24 h (agudas), o las que se presenten desde 24 h hasta 14 días después de la infusión (tardías).
En el presente estudio nos centramos en los AA agudos, que se presentan durante la administración del fármaco o en las 24 h posteriores, en enfermos de AR en condiciones de práctica clínica. Los AA agudos que se observaron fueron prurito, edema, urticaria, hipotensión, bradicardia, taquicardia, cefalea, fiebre o choque anafiláctico extremo, entre otras.
El registro BIOBADASER (Registro de Acontecimientos Adversos de Terapias Biológicas), creado en 2001 para determinar la seguridad de las terapias biológicas en enfermedades reumáticas, ofrece datos de seguridad, AA relevantes y tasas de retención5. Comparativamente, observamos un total de 12 AA en 7 pacientes de los 111 incluidos, 9 de los cuales fueron en el momento de la administración del tratamiento y 3 en las 24 h posteriores. Estos datos corresponden a un 10,8% de los pacientes con AA, un 8,1% durante la administración y un 2,7% en las siguientes 24h. En el registro BIOBADASER5 se informa un total de 761 reacciones asociadas al tratamiento; teniendo en cuenta que, a noviembre del 2009, se han registrado 5.493 pacientes, corresponde a un porcentaje del 13,8% de pacientes con AA asociados al tratamiento. Si se compara estos resultados con los registrados en BIOBADASER, se observa que aunque la presente muestra es menor, el porcentaje de AA no difiere del registrado en BIOBADASER. Hay que tener en cuenta que en el porcentaje de BIOBADASER se incluyen también aquellos AA observados en fármacos de administración por vía subcutánea, aunque en un estudio realizado por el grupo BIOBADASER en 20088 se pone de manifiesto que la frecuencia de aparición de reacciones adversas es mayor con infliximab que con los otros fármacos anti-TNF-α de administración por vía subcutánea. Igualmente, en estudios realizados con rituximab9 y abatacept10, los AA relacionados con el tratamiento oscilan del 10 al 15% de los pacientes.
Respecto de la frecuencia de AA relacionados con el fármaco administrado en nuestra muestra, observamos que es superior en el tratamiento con infliximab respecto de los otros tratamientos, explicado por el tamaño de la muestra que recibe este tratamiento (73% de pacientes) respecto del 20,7% que recibe abatacept y el 13,5% que recibe rituximab. Dos pacientes con rituximab presentaron AA durante la administración y ninguno de los tratados con abatacept. Ninguno de los AA registrados fue grave; únicamente uno fue calificado de intensidad moderada (urticaria) y el resto, de intensidad leve. En la mayoría de los artículos publicados, el porcentaje de AA graves es también bajo, pero en los fármacos de administración por vía intravenosa la reacción es, en algunos casos, la causa de interrupción del tratamiento, a pesar de que en algunas ocasiones se controlan con medicación que reduzca la sensibilización o disminuyendo el ritmo de infusión.
En nuestros casos se estableció una relación definitiva del AA con la medicación; en 2 casos se estableció una relación probable y en el caso restante la relación establecida fue posible. Tres de los casos se resolvieron con la interrupción temporal del tratamiento; en un caso se ajustó la dosis y en los 3 restantes no se emprendió ninguna acción. Respecto de la aparición del AA durante la administración del tratamiento o en las 24h siguientes, observamos que en el 71,5% de los pacientes la reacción se produce durante el tratamiento y en el 28,5% restante se produce en las siguientes 24 h.
El presente trabajo pretende resaltar datos fundamentales en el manejo de las terapias biológicas. Primero, se debe destacar que el tipo de reacciones observadas relacionadas con los tratamientos de administración por vía intravenosa es similar al descrito anteriormente en diferentes estudios para el uso de estos tratamientos biológicos. Segundo, no hemos observado ningún AA grave, aunque es importante estar alerta ante la posible aparición de estos AA durante y 24 h tras la infusión, ya que pueden ser motivo de interrupción del tratamiento. Destaca también la alta prevalencia de tratamiento con corticoides de los pacientes con AR, no siempre registrado, así como la tasa de vacunación antigripal y antineumocócica baja, a pesar de las recomendaciones existentes. Por último, se debe destacar que no solo durante la infusión se debe monitorizar la administración, sino que enfermería desempeña un papel destacado en el control posterior durante las 24 h siguientes a la administración.
Responsabilidades éticasProtección de personas y animalesLos autores declaran que para esta investigación no se han realizado experimentos en seres humanos ni en animales.
Confidencialidad de los datosLos autores declaran que han seguido los protocolos de su centro de trabajo sobre la publicación de datos de pacientes y que todos los pacientes incluidos en el estudio han recibido información suficiente y han dado su consentimiento informado por escrito para participar en dicho estudio.
Derecho a la privacidad y consentimiento informadoLos autores han obtenido el consentimiento informado de los pacientes y/o sujetos referidos en el artículo. Este documento obra en poder del autor de correspondencia.
Conflicto de interesesEl presente trabajo multicéntrico ha sido realizado con la colaboración en igual medida de todos sus autores. Roche Pharma ha colaborado desinteresadamante en la gestión de datos y logística. No ha participado en la elaboración del documento. No existe financiación para elaboración del proyecto.


