



El raquitismo hipofosfatémico ligado al cromosoma X (XLH) es la principal forma de raquitismo hereditario causada por la mutación del gen PHEX y que se manifiesta principalmente en la infancia. Clínicamente cursa con retraso en el crecimiento y deformidades óseas, sin embargo, existen formas de presentación atípicas que dificultan el diagnóstico. Presentamos un caso de XLH con diagnóstico tardío y forma paucisintomática que presenta múltiples fracturas y gran afectación en su calidad de vida, en tratamiento con la terapia clásica para esta enfermedad.
X-linked hypophosphataemic rickets (XLH) is the main form of hereditary rickets caused by mutation of the PHEX gene and occurs mainly in childhood. Clinically, it causes growth retardation and bone deformities; however, there are atypical forms of presentation that make diagnosis difficult. We present a case of XLH of late diagnosis and paucisymptomatic form with multiple fractures and greatly affecting quality of life, under treatment with traditional therapy for this disease.
El raquitismo hipofosfatémico ligado al cromosoma X (XLH) es una enfermedad hereditaria causada por la mutación del gen PHEX localizado en el locus Xp22.111. Este gen codifica para una endopeptidasa reguladora de fosfatos cuya función es inhibir el factor de crecimiento fibroblástico 23 (FGF-23). El aumento de este disminuye la reabsorción tubular de fosfato (RTP) y la actividad de alfa-1-hidroxilasa, produciendo disminución en los niveles séricos de 1,25-dihidroxivitamina D, hiperfosfaturia e hipofosfatemia2.
Los principales hallazgos clínicos incluyen: raquitismo, osteomalacia, retraso del crecimiento, dolores óseos y entesopatías, sin embargo, hay formas paucisintomáticas que solo se manifiestan con dolor crónico, osteoartritis y debilidad muscular, lo que dificulta y retrasa su diagnóstico, precisando un manejo multidisciplinar3. El tratamiento se basa en suplementos de fósforo y calcitriol; sin embargo, la aparición del nuevo anticuerpo monoclonal humano anti-FGF-23 permite bloquear el mecanismo de la enfermedad y modificar su historia natural.
Caso clínicoVarón de 35 años con antecedente de dolor lumbar crónico invalidante y mialgias de varios años de evolución, sin signos de osteoartritis ni síntomas constitucionales, que es remitido para valoración por reumatología. La exploración física era normal, talla de 185cm y TA de 145/85mmHg. En las pruebas complementarias solicitadas destacaron: fósforo 1,7mg/dl (rango normal [RN]: 2,7-4,5mg/dl), hormona paratiroidea 85,2pg/ml (RN: 15-65pg/ml), 1,25 dihidroxivitamina D 18,7ng/ml (RN: ≥30ng/ml), creatinina 0,9mg/dl (RN: 0,5-0,95mg/dl), fosfatasa alcalina 167U/l (RN: 35-105U/l), fosfaturia 2.088mg/24h con RTP del 55% (RN: ≥80%). El resto de resultados fueron normales.
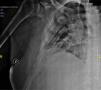
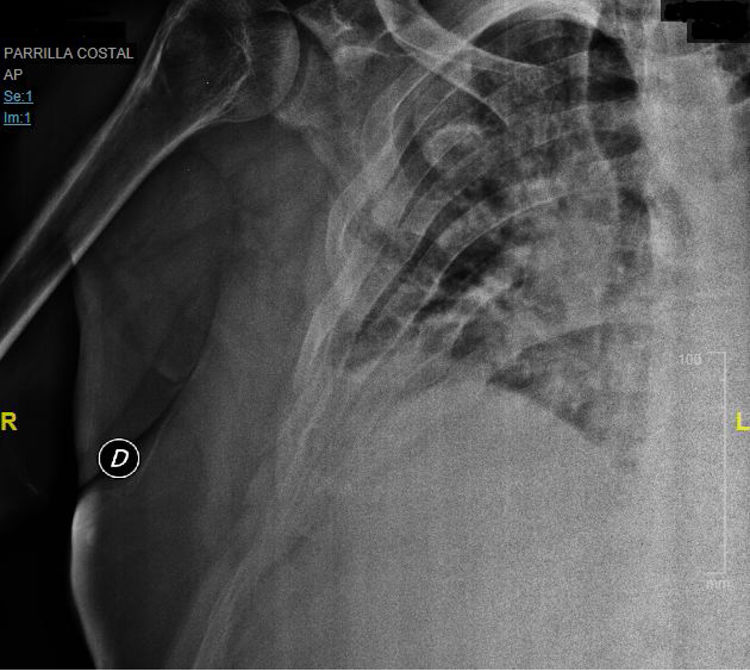
Presentaba múltiples fracturas en costillas y escápula derecha debido a caída casual (fig. 1) y una densitometría con signos de osteopenia en columna vertebral. Ante estos hallazgos se inició tratamiento con fosfato, calcitriol y analgésicos.
Se descartó osteomalacia tumoral como posible causa, por lo que se realizó estudio genético que confirmó el diagnóstico de XLH con mutación en el gen PHEX (4c.1645t). Actualmente se mantiene el tratamiento con aportes de fósforo y calcitriol, pero persiste el dolor lumbar con importante afectación en su calidad de vida. Por ello, este paciente sería un buen candidato para beneficiarse del uso de burosumab.
DiscusiónEl diagnóstico de las enfermedades raras constituye un reto, en especial el XLH, cuya expresión fenotípica es muy variable, si bien la mayor parte de los casos son diagnosticados durante la infancia, este caso es un ejemplo de una forma de presentación poco expresiva que genera un amplio diagnóstico diferencial que podría implicar la participación de diversos especialistas.
Las pruebas genéticas ayudan en aquellos casos con presentaciones atípicas o en la edad adulta en el que la osteomalacia tumoral es la principal posibilidad. El tratamiento clásico con fosfato y calcitriol lleva a efectos secundarios que limitan la adherencia al tratamiento a largo plazo, pero el tratamiento con burosumab ha demostrado resultados muy positivos tanto en población infantil como en adultos4,5. Este se ha relacionado con la mejoría clínica, la corrección de las deformidades, la normalización del crecimiento y de los niveles de fósforo de forma mantenida, con buenos resultados también en pacientes que, aun habiendo finalizado su crecimiento, persisten con importante limitación en su calidad de vida por las secuelas de la enfermedad6,7.
En conclusión, debemos estar alerta de estos casos de XLH con diagnóstico complicado por su presentación paucisintomática que pueden remitirnos sin diagnóstico ya en edad adulta. Burosumab podría ser de gran utilidad en el tratamiento de pacientes adultos que presenten importante limitación en su calidad de vida.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.


