







Las miopatías inflamatorias son un grupo heterogéneo de miopatías en las que existe inflamación en la biopsia. En su evaluación es esencial el uso de técnicas neurofisiológicas que permiten obtener información sobre la naturaleza del proceso.
Este trabajo revisa el patrón electromiográfico característico de las miopatías inflamatorias, su valor diagnóstico, las limitaciones y algunas pistas sobre la interpretación de los resultados de las técnicas neurofisiológicas en la evaluación de las miopatías inflamatorias.
Inflammatory myopathies are a heterogeneous group of myopathies in which there is biopsy-evident inflammation. In its evaluation it is essential to use neurophysiological techniques that provide information on the nature of the process.
This paper reviews the electromigram pattern characteristic of inflammatory myopathies, its diagnostic value, limitations, and some clues on the interpretation of the results of neurophysiological techniques in the assessment of inflammatory myopathies.
Las miopatías inflamatorias (MI) son un grupo heterogéneo de enfermedades musculares autoinmunitarias en el que se incluyen la polimiositis, la dermatomiositis y la miositis con cuerpos de inclusión (MCI)1–4. Pueden presentarse de forma aislada o asociadas a otras enfermedades autoinmunes y cursan generalmente con elevación de enzimas musculares y pérdida de fuerza simétrica de predominio proximal.
Aunque la MCI parece ser una entidad clínicamente definida con una patogenia eminentemente degenerativa, se incluye tradicionalmente en las sucesivas clasificaciones de MI, por presentar también inflamación en la biopsia5.
El diagnóstico de las MI se basa en la historia clínica, la exploración física, la elevación de proteínas musculares en sangre, la determinación de anticuerpos más o menos específicos, las pruebas neurofisiológicas, las pruebas de imagen y el estudio anatomopatológico del músculo obtenido mediante biopsia.
Las pruebas neurofisiológicas incluyen los estudios de electroneurografía o de conducción nerviosa y el electromiograma de aguja (EMG) y permiten explorar el funcionamiento del sistema nervioso periférico, incluyendo nervio, unión neuromuscular y músculo. La utilidad de los estudios neurofisiológicos radica en su alta sensibilidad y especificidad para el diagnóstico de las miopatías y en la capacidad de descartar otras causas de debilidad como son neuropatías con afectación motora, síndromes miasténicos o enfermedades de motoneurona.
Como regla general, el estudio electromiográfico suele empezar a mostrar información al cabo de unas 2-3 semanas de iniciarse el proceso patogénico, por lo que, en miopatías agudas, se recomienda realizar el estudio en unas 3 semanas desde el inicio de los síntomas como pronto, para asegurar una buena sensibilidad del estudio.
La sensibilidad de las pruebas neurofisiológicas en las MI no ha sido objeto de muchos estudios. En el clásico trabajo de Bohan y Peter, la sensibilidad de estas fue del 89%6. Por tanto, debe recordarse que el EMG puede ser normal, dependiendo de la exhaustividad del estudio, el número de músculos explorados, el momento de realización, la capacidad de colaboración del paciente, la experiencia del electromiografista, etc.
El EMG permite seleccionar el músculo que se va a biopsiar. Como la aguja de electromiografía lesiona el tejido muscular, no es aconsejable biopsiar un músculo estudiado mediante dicha técnica. Nosotros generalmente solemos realizar el EMG sobre musculatura clínicamente débil y biopsiar en el lado contralateral al músculo que muestra alteraciones eléctricas. La correlación entre los hallazgos electromiográficos en una miopatía y los histopatológicos es alta, entre un 70-90%7.
La realización del EMG puede aumentar discretamente los niveles sanguíneos de creatinfosfocinasa (CPK) y de otras enzimas musculares. Por ello es conveniente, si es necesario realizar una determinación de CPK, hacer una extracción antes de la realización del EMG o al menos 3 días después del EMG. Si se hiciera inmediatamente después conviene tener en cuenta la posible elevación de enzimas musculares debida a la lesión traumática con la aguja8.
Ni la antiagregación ni la anticoagulación se consideran contraindicación absoluta para la realización de un EMG.9,10. No obstante, en los pacientes anticoagulados en exceso o con trastornos graves de la coagulación, debe sopesarse la relación riesgo-beneficio y evitar el estudio de músculos profundos o de difícil compresión en caso de hemorragia.
Protocolo neurofisiológico para el estudio de las miopatías inflamatoriasEn general se debe estudiar: al menos una conducción motora y sensitiva de un miembro afecto, y electromiografía de aguja de músculos afectados, por lo menos uno proximal y otro distal11. Es también recomendable realizar un estudio de estimulación repetitiva en aquellos casos en los que se sospeche un trastorno de la unión neuromuscular.
Se puede estudiar cualquier músculo, pero es recomendable seguir una sistemática racional. Se deben explorar músculos clínicamente débiles y de las extremidades superiores e inferiores. Si es necesario, se explorarán músculos paraespinales o bulbares.
En general, los hallazgos electroneurográficos en una miopatía mostrarán una conducción nerviosa normal, con una posible disminución de la amplitud de los potenciales motores cuando la miopatía sea distal, grave y esté evolucionada.
El EMG es la parte más importante del estudio neurofisiológico. Permite estudiar la actividad eléctrica muscular mediante un electrodo concéntrico de aguja insertado en el músculo que detecta diferencias de potencial extracelular. El estudio del músculo se hace en 2 situaciones: en reposo y en actividad voluntaria.
En reposo se valora la denominada actividad espontánea. En el músculo normal no debe existir actividad espontánea, salvo el denominado ruido de placa. Se trata de un sonido como el de una caracola al ponerla en el oído y representa la actividad de despolarización de la placa motora por la liberación basal de acetilcolina.
La actividad espontánea anormal o patológica puede ser de varios tipos:
- 1.
Fibrilaciones y ondas positivas.
- 2.
Descargas de alta frecuencia.
- 3.
Descargas miotónicas.
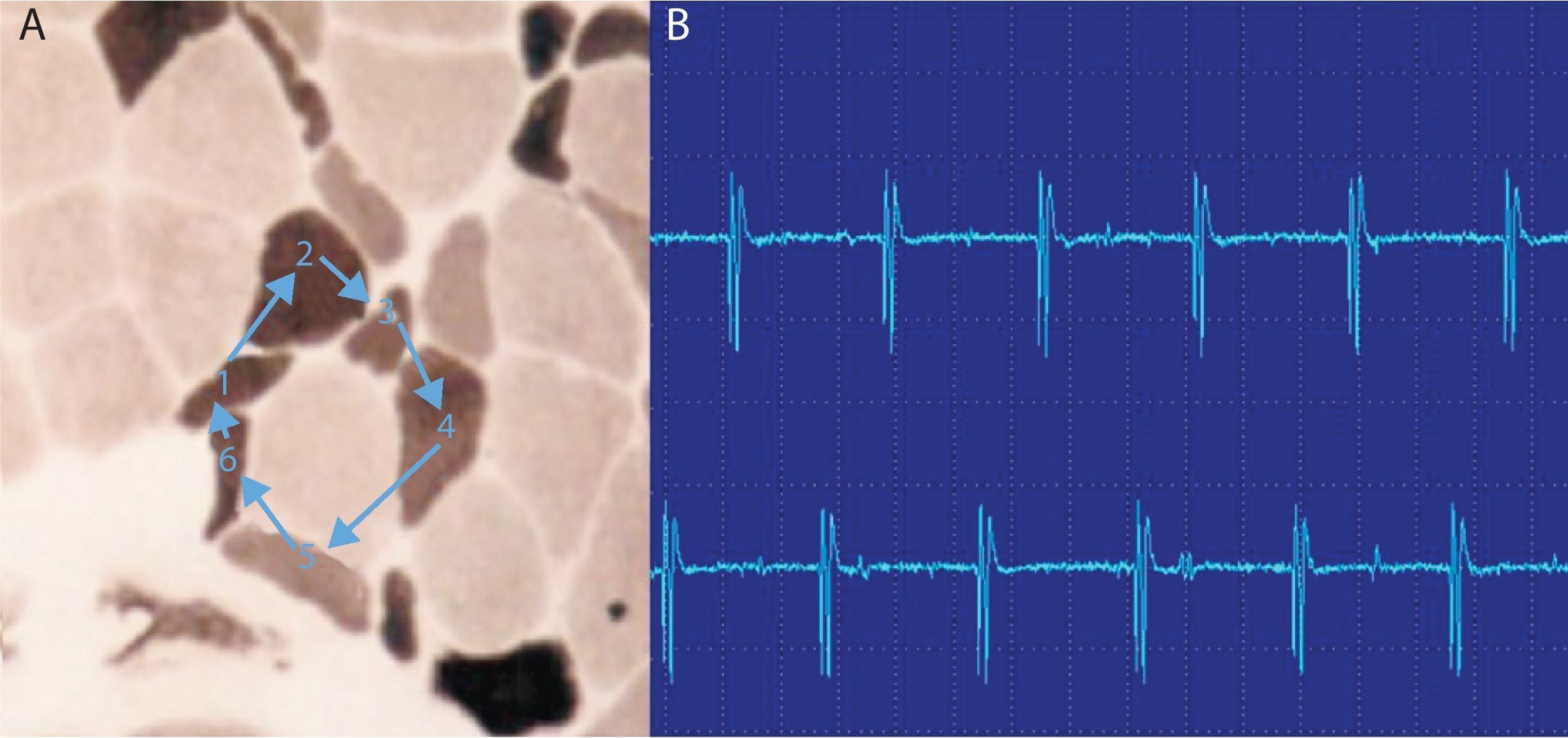
Las fibrilaciones y ondas positivas (figs. 1 y 2) son pequeños potenciales eléctricos, siempre de inicio positivo, siempre rítmicas, que se deben a la generación de potenciales de acción en las fibras musculares aisladas denervadas. Son típicas de los procesos denervativos, pero también se pueden detectar en las miopatías con necrosis o inflamación importante. Se cree que en estas miopatías los fenómenos de necrosis de la fibra muscular son tan importantes que producen la denervación de algunas fibras.
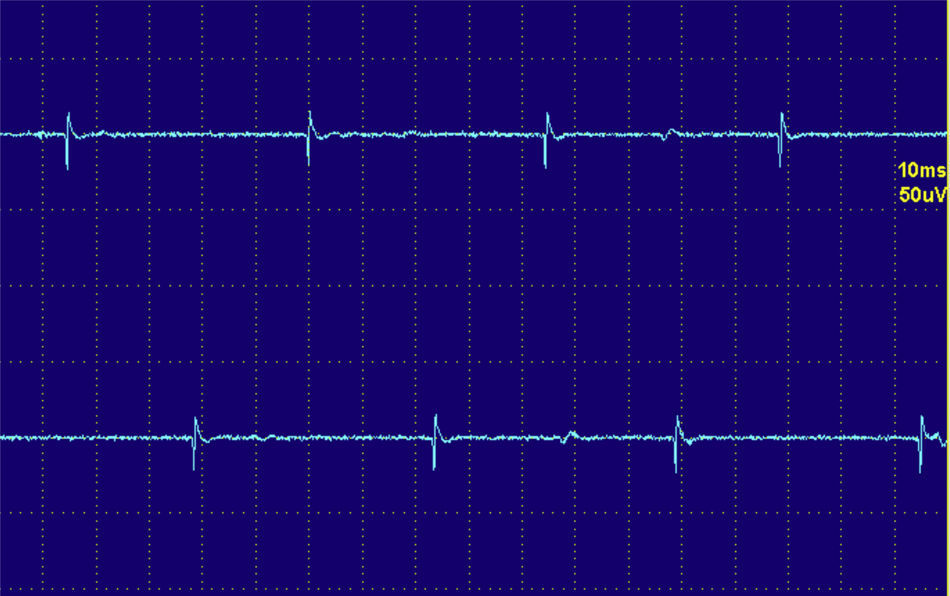
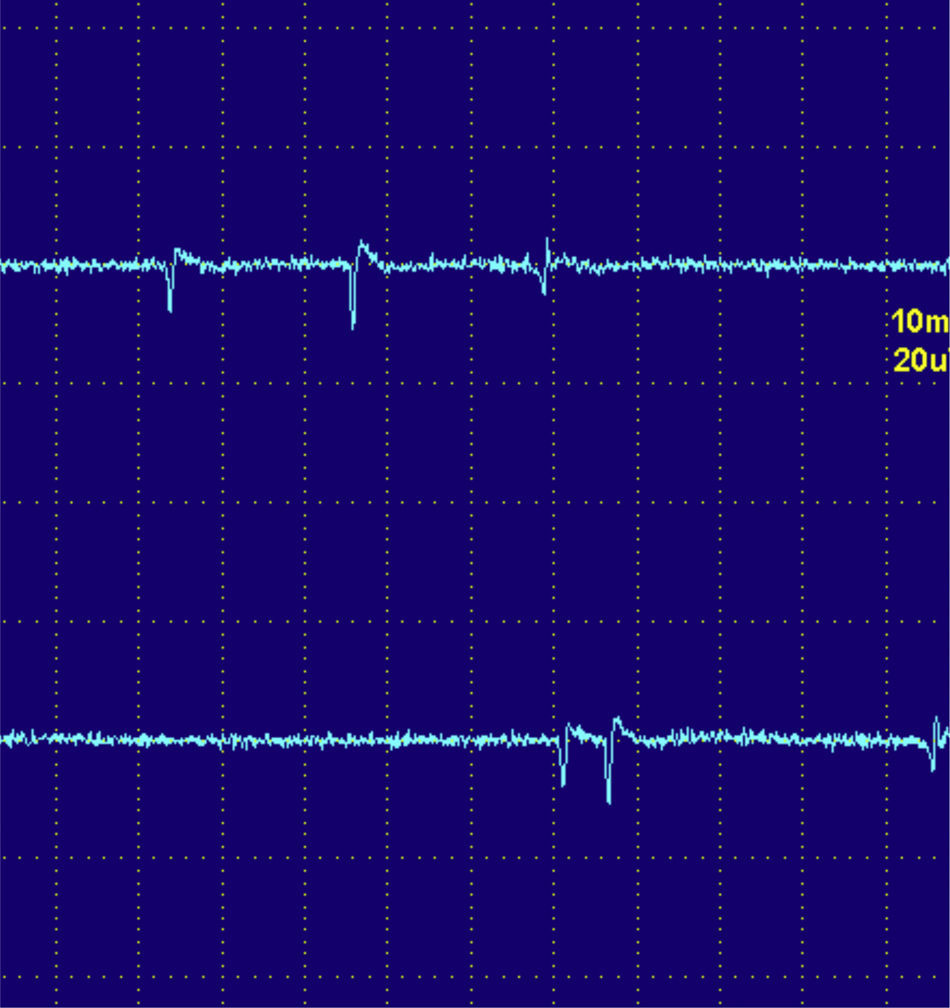
Las descargas de alta frecuencia, también llamadas pseudomiotónicas o descargas repetitivas complejas, tienen un inicio y un fin brusco y suenan como una maquinaria con una cadencia rítmica o pseudomiotónica. Se producen por transmisión efáptica, es decir, por contigüidad, de un potencial generado espontáneamente. Son también típicas de procesos denervativos de larga evolución, y, al igual que las fibrilaciones y ondas positivas, pueden encontrarse en las MI (fig. 3)12.
Descargas repetitivas de alta frecuencia. A) El potencial se origina espontáneamente en la fibra muscular número 1 y por transmisión efáptica, por contigüidad, pasa a la 2, y sucesivamente a la 3, 4, 5, 6 y de nuevo a la 1. B) El electromiograma recoge un potencial polifásico que se inicia y acaba bruscamente, que es siempre el mismo y dispara rítmicamente, y que tiene característicamente un sonido como el de una máquina, «chaca-chaca-chaca».
Las descargas miotónicas son la representación electrofisiológica de la dificultad para la relajación que sigue a la contracción muscular voluntaria o inducida, generalmente por percusión, conocida como miotonía. Son debidas a la hiperexcitabilidad transitoria de la membrana de la fibra muscular. La miotonía se identifica clínicamente con facilidad, por la característica dificultad y lentitud para la relajación muscular tras una contracción mantenida durante unos segundos. El fenómeno miotónico disminuye en intensidad tras varias contracciones repetidas. Desde un punto de vista electrofisiológico refleja la actividad de fibras musculares individuales que descargan de manera espontánea y repetitiva. Tiene un sonido característico semejante a la caída de un avión en picado, y una vez identificado es fácilmente reconocible. Las descargas miotónicas son típicas de la distrofia miotónica, pero pueden observarse aisladamente en las MI. Son especialmente frecuentes en la miopatías por antimaláricos.
Durante la actividad voluntaria se pide al paciente que realice un esfuerzo leve, a fin de poder observar los potenciales de unidad motora aislados (PUM) y poder analizarlos adecuadamente. También se analiza la activación y el reclutamiento de las fibras musculares, es decir, la manera que tiene el músculo de aumentar su capacidad de contracción. Para ello se suele pedir al paciente que haga el máximo esfuerzo con ese músculo, generalmente contra resistencia.
Los PUM son la sumación de los potenciales de acción de las fibras musculares inervadas por una motoneurona, es decir, son el reflejo neurofisiológico de la unidad motora. Se recomienda analizar unos 20 PUM. Se estudia la amplitud, duración y la cantidad de veces que el potencial cruza la línea de base (relacionados directamente con la sincronía en la contracción de las fibras), lo que se denomina fase. Aquellos PUM con 5 fases o más se denominan polifásicos.
En las fases agudas de una miopatía existe una pérdida de miofibrillas y de sincronía en la contracción. Por ese motivo los PUM miopáticos son pequeños en amplitud, breves en duración y polifásicos (fig. 4).
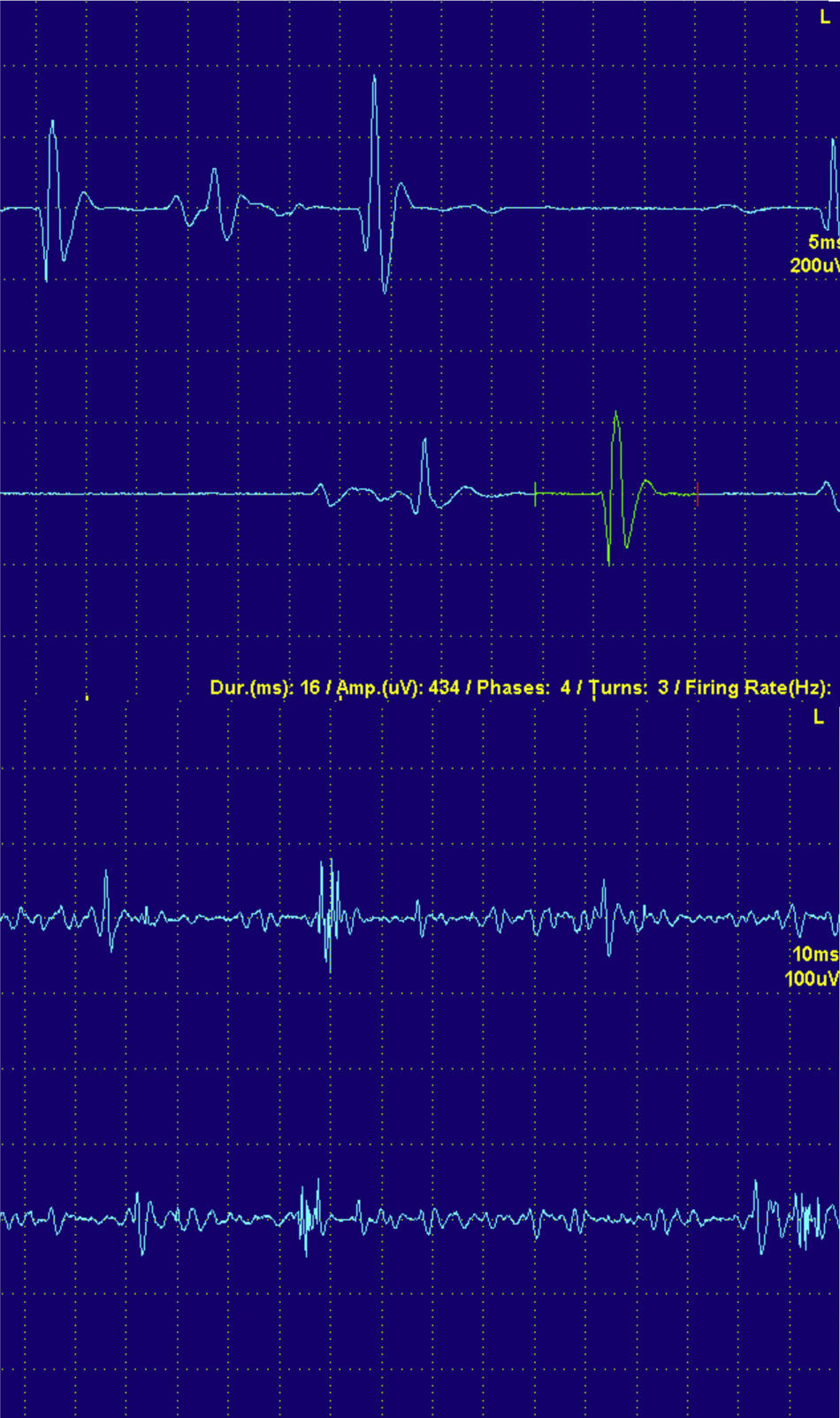
En las fases más crónicas de la miopatía y especialmente en la MCI, como el proceso es más crónico, es habitual que los PUM sean de mayor tamaño y también de mayor duración. Estos PUM reflejan la regeneración del músculo en unidades con mayor número de fibras musculares.
En el análisis del patrón de reclutamiento al máximo esfuerzo, se suele observar un patrón de reclutamiento precoz, es decir, de un mayor número de unidades motoras que las que se suelen reclutar para un esfuerzo relativamente submáximo; el paciente tiene que reclutar más unidades motoras para obtener una fuerza relativamente baja13.
El EMG en las MI suele mostrar lo que se define habitualmente como «patrón neurógeno-miopático» o «patrón mixto»: actividad espontánea con PUM pequeños, cortos y polifásicos, y reclutamiento precoz. Este patrón es altamente sugestivo, pero no específico, de una MI. Puede observarse también en algunas miopatías con importante destrucción muscular como la distrofia facioescapulohumeral, algunas distrofias de cinturas, o en las distrofinopatías.
Para un electromiografista es relativamente sencillo detectar actividad espontánea. Es algo más difícil detectar los PUM miopáticos, especialmente en pacientes con miopatías leves. No se debe olvidar que la interpretación de un EMG es generalmente subjetiva y dependiente de observador14.
Los hallazgos electromiográficos en una miopatía pueden considerarse relativamente inespecíficos, si bien la presencia de un patrón mixto sugiere, como se ha comentado, una MI. Aunque se han descrito patrones propios de cada una de las 3 entidades nosológicas tradicionales15, el diagnóstico definitivo suele hacerse siempre con la biopsia muscular2.
Aunque se ha utilizado el EMG para monitorizar la respuesta al tratamiento y la evolución de una miopatía, nosotros, como otros autores16, preferimos emplear la exploración física y los niveles de CPK, especialmente en la polimiositis.
Nuevas técnicas: análisis de turns-amplitud o de giros-amplitudEste tipo de estudio se basa en un análisis matemático del electromiógrafo, que mide la relación entre los giros y la amplitud de los PUM y los representa en una gráfica. Esta técnica permite diferenciar procesos miopáticos de los neurógenos. En los primeros este cociente es menor que en los segundos. El estudio suele hacerse en el patrón de máximo esfuerzo17.
Diagnóstico diferencialEn el estudio de las MI existen diversas situaciones en las que se plantean diagnósticos diferenciales con otras entidades nosológicas y que tienen importantes implicaciones diagnósticas, pronósticas y de tratamiento. No se debe olvidar que el diagnóstico de una miopatía inflamatoria solo puede hacerse con certeza con una biopsia muscular18.
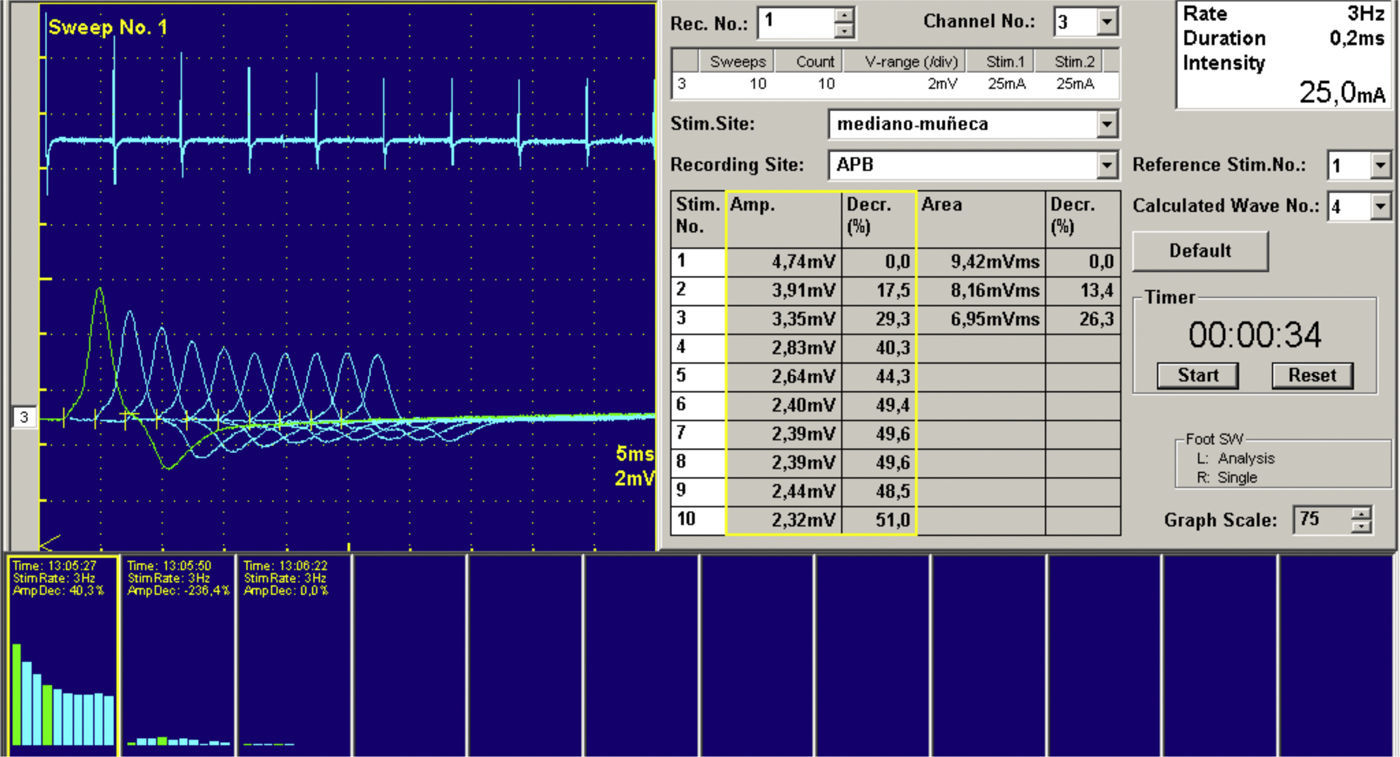
«Pseudopolimiositis» (tabla 1): algunas miopatías hereditarias, como la distrofia facioescapulohumeral o la disferlinopatías pueden presentarse de manera aguda o subaguda con elevaciones de CPK, y pueden llevar al diagnóstico erróneo de una MI19. Algunas de estas enfermedades presentan una herencia autosómica recesiva y la historia familiar no necesariamente ayuda. El diagnóstico puede complicarse incluso con la biopsia, ya que alguna de estas entidades pueden tener infiltrados inflamatorios en el músculo. El EMG no suele ayudar en estos pacientes, puesto que los hallazgos son similares. Algunos detalles clínicos, como la debilidad facial de una distrofia fascioescapulohumeral o la afectación distal en las disferlinopatías pueden dar pistas sobre el diagnóstico. La ayuda de un neurólogo experto en enfermedades neuromusculares puede ser de gran interés para el reumatólogo. Otras miopatías que pueden presentarse como «pseudopolimiositis» son las metabólicas, como la enfermedad de McArdle o las miopatías mitocondriales, la enfermedad de Pompe, las miopatías parasitarias o las tóxicas, que merecen un apartado propio. Hay otros simuladores de MI, como pueden ser neuropatías o enfermedades de la unión neuromuscular, como una miastenia gravis. Existe también la posibilidad de que alguna de estas enfermedades se combinen en un mismo paciente, y aunque esto sea poco frecuente, debe tenerse en cuenta cuando el tratamiento de la MI no esté resultando eficaz. Como hemos comentado previamente, el EMG no siempre es capaz de aportar datos en el diagnóstico diferencial. En síndromes miasteniformes, como la miastenia gravis, alguna miastenia congénita o el síndrome de Lambert-Eaton, el EMG permite, mediante las técnicas de estimulación repetitiva, ofrecer el diagnóstico con una alta especificidad (fig. 5).
Falsas miopatías inflamatorias
| Fibromialgia |
| Polimialgia reumática |
| Miositis granulomatosas |
| Miopatía por hipotiroidismo |
| Miopatías metabólicas |
| Glucogenosis (enfermedad de McArdle, enfermedad de Pompe) |
| Del metabolismo lipídico |
| Mitocondriales |
| Miopatías hereditarias |
| Distrofinopatías |
| Disferlinopatías |
| Distrofia facioescapulohumeral |
| Déficit de merosina |
| Distrofia de cinturas |
| Miopatías farmacológicas |
Miopatías tóxicas: es necesario recordar que no todos los cuadros de pérdida de fuerza proximal se deben a una miopatía inflamatoria. Existen otras muchas formas de miopatía, farmacológicas (tabla 2), tóxicas20, infecciosas, hereditarias, metabólicas, etc., que pueden manifestarse clínicamente de manera indistinguible de una MI. En estas enfermedades el EMG no suele ofrecer tampoco ayuda en la diferenciación, sino que es la historia cuidadosamente recogida, la retirada del tóxico y la posterior mejoría del cuadro clínico la que suele permitir el diagnóstico.
Miopatías farmacológicas
| Hipolipemiantes |
| Estatinas |
| Ezetimiba |
| Fármacos que aumentan el riesgo de miopatía por estatinas |
| Amiodarona |
| Fibratos |
| Niacina |
| Ciclosporina |
| Imidazoles |
| Macrólidos |
| Verapamilo |
| Fármacos usados frecuentemente en reumatología |
| D-penicilamina |
| Colchicina |
| Cloroquina e hidroxicloroquina |
| Interferón alfa |
| Ciclosporina |
| Tacrólimus |
| Esteroides |
| Germanio |
Algunas miopatías yatrógenas tienen un interés particular para el reumatólogo al ser secundarias a fármacos usados en el tratamiento de otras enfermedades reumatológicas. Un caso particular es la miopatía por colchicina21, La colchicina es un fármaco antimicrotúbulos que se une a la tubulina y evita la polimerización y la formación de microtúbulos22. Produce una miopatía que puede ser aguda y potencialmente letal. También puede originar una neuropatía asociada. Al ser un potente inhibidor del citocromo P450 CYP3A4, es capaz de potenciar la acción miotóxica de otros fármacos, como las estatinas.
En la miopatía por colchicina, el cuadro clínico puede ser superponible al de una MI convencional, con la salvedad de que suele existir una hiporreflexia desproporcionada a la pérdida de fuerza y una pérdida leve de la sensibilidad vibratoria, aunque estos son signos de la exploración muy sutiles y que pueden pasarse por alto fácilmente. La CPK puede estar levemente elevada. En este cuadro, el EMG es especialmente útil, puesto que además de un patrón mixto, puede detectarse miotonía eléctrica. Ocasionalmente se observa también clínicamente23. Además el EMG permite estudiar la neuropatía asociada, lo que también apoya el diagnóstico. El tratamiento consiste en retirar la colchicina.
La cloroquina y la hidroxicloroquina pueden también producir miopatía24. En general se trata de una miopatía leve con elevaciones discretas de CPK y mayores de LDH, aunque se han descrito casos de mayor gravedad25,26. El EMG suele mostrar un patrón miopático inespecífico en un 53% de los casos24. En caso de sospecha debe retirarse el fármaco si la miopatía es clínicamente importante.
Recaída vs miopatía esteroidea. Una situación relativamente frecuente con la que se encuentra el clínico es el paciente que se queja de un empeoramiento de la fuerza, sobre todo proximal, de las extremidades inferiores. Cuando esto ocurre suele existir la duda de si el cuadro se trata de una recaída de la polimiositis o de una miopatía esteroidea. En este escenario el EMG sí puede ser de ayuda, combinado con la exploración y otras pruebas complementarias. El EMG en las recaídas suele mostrar signos de destrucción activa del músculo, es decir, actividad espontánea4,27. Sin embargo, en la miopatía esteroidea, el EMG suele ser normal. En la miopatía esteroidea, los niveles de CPK también suelen haberse normalizado, o al menos mejorado, y tienden a aumentar en las recaídas. Ocasionalmente es necesario realizar una nueva biopsia muscular para asegurarse el diagnóstico.
Conclusiones: el diagnóstico de una MI puede ser difícil. Junto con los hallazgos clínicos y de las pruebas de laboratorio, las pruebas neurofisiológicas permiten al clínico apreciar la riqueza de la fisiología y la fisiopatología del sistema neuromuscular. Conocer cuándo y qué músculos explorar neurofisiológicamente y cómo interpretar los hallazgos del EMG es esencial para realizar un diagnóstico y un manejo correctos de las MI.
Responsabilidades éticasProtección de personas y animales. Los autores declaran que para esta investigación no se han realizado experimentos en seres humanos ni en animales.
Confidencialidad de los datos. Los autores declaran que en este artículo no aparecen datos de pacientes.
Derecho a la privacidad y consentimiento informado. Los autores han obtenido el consentimiento informado de los pacientes y/o sujetos referidos en el artículo. Este documento obra en poder del autor de correspondencia.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.


