




La asociación de dermatomiositis y miastenia gravis (MG) es infrecuente, habiéndose comunicado hasta la actualidad únicamente 26 casos. Se presenta el caso de un varón de 69 años diagnosticado de MG 2 años atrás, en tratamiento con piridostigmina, que inicia cuadro agudo de debilidad muscular proximal, artralgias en hombros y elevación de creatincinasa (CK); así como aparición de eritema facial generalizado y pápulas de Gottron. En el estudio de laboratorio se evidenció positividad de anticuerpos antinucleares y anti-Mi2. Ulteriores determinaciones de CK mostraron niveles por encima de 1.000U/l. Se discute el manejo clínico de este paciente y las implicaciones terapéuticas que plantea la coexistencia de ambas entidades.
The association of dermatomyositis with myasthenia gravis (MG) is uncommon, having been reported so far in only 26 cases. We report the case of a 69 year-old man diagnosed with MG two years ago and currently treated with piridostigmyne. The patient developed acute proximal weakness, shoulder pain and elevated creatine-kinase (CK). He also developed generalized facial erythema and Gottron's papules. Laboratory tests showed positive antinuclear and anti-Mi2 antibodies. Further analysis confirmed CK levels above 1000U/l. The clinical management of the patient and the therapeutic implications derived from the coexistence of both entities are discusssed.
Las miopatías inflamatorias idiopáticas (MII), entre las que se incluye la dermatomiositis (DM), son un grupo heterogéneo de enfermedades autoinmunes sistémicas cuya principal característica clínica es la debilidad muscular de predominio proximal1. Son enfermedades infrecuentes, con una incidencia anual estimada de 6 casos/105 individuos2, pudiendo asociarse a otras enfermedades autoinmunes, como el lupus o el síndrome de Sjögren.
Otra enfermedad autoinmune que cursa con debilidad muscular, la miastenia gravis (MG), también puede asociarse a las MII3–9. La MG es infrecuente, con una prevalencia estimada de 140 casos/106 habitantes10. Su patogenia deriva del compromiso de la transmisión neuromuscular, resultante de la unión de autoanticuerpos a proteínas implicadas en su señalización. Reconocer la asociación entre ambas entidades es importante por sus implicaciones terapéuticas. Presentamos un caso de DM en un paciente previamente diagnosticado de MG.
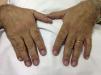
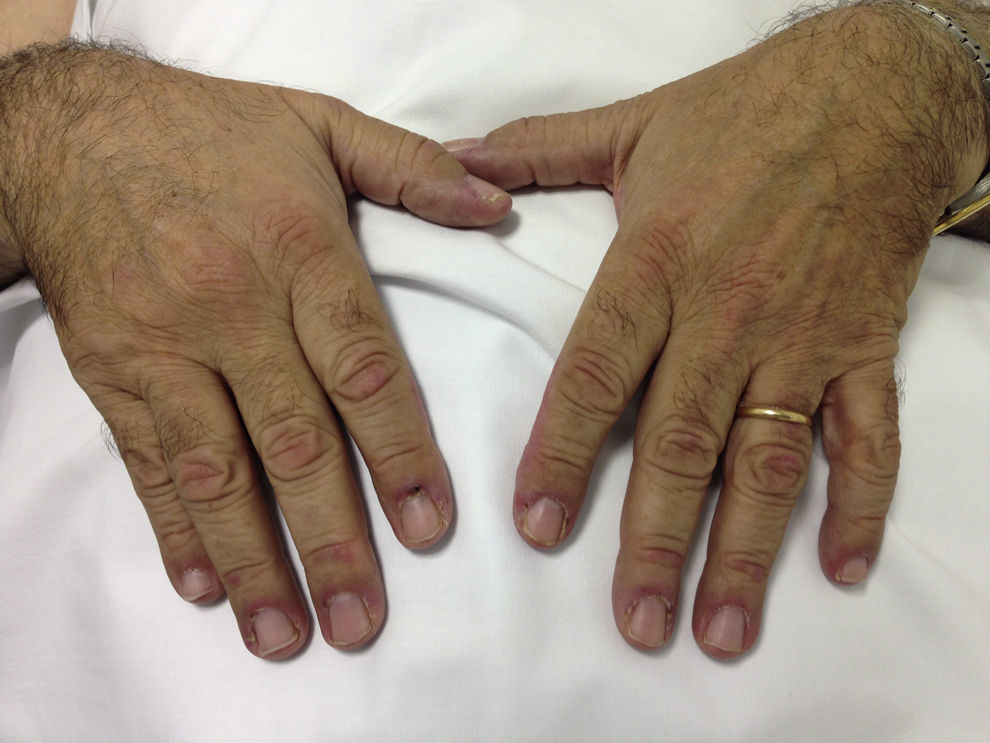
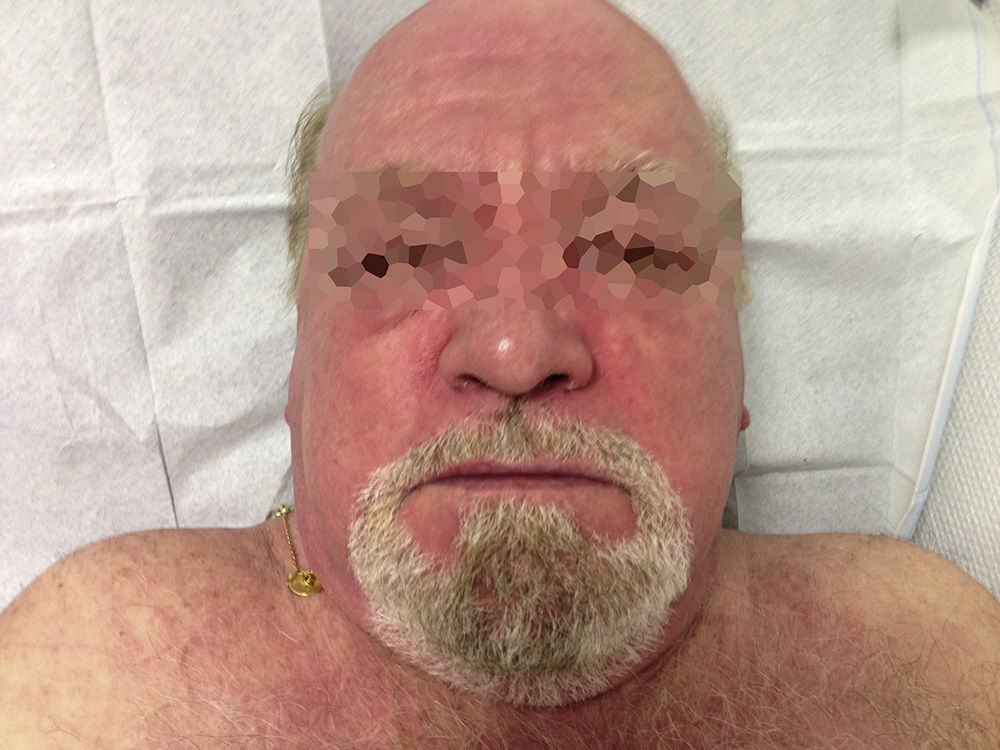
Caso clínicoVarón de 69 años de raza blanca, diagnosticado 2 años atrás de MG, con anticuerpos anti-AChR positivos y en tratamiento con piridostigmina (60mg/8h). Fue remitido por la aparición de debilidad muscular desde hacía un mes, con dificultad para vestirse o levantarse de la cama, artralgias en hombros y elevación de creatincinasa (CK). Presentaba eritema facial difuso desde hacía 6 meses. La exploración física mostró engrosamiento cuticular con hemorragias, pápulas de Gottron (fig. 1) y eritema facial, en chal y en escote (fig. 2). Presentaba debilidad de la musculatura proximal en miembros superiores e inferiores (4/5). El resto de la exploración general fue normal.
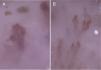
En el estudio analítico destacaba una CK de 1.254U/l. Los anticuerpos antinucleares fueron positivos (>1/160, patrón granular; anti-ENA y anti-DNA negativos), así como los anti-Mi2. La capilaroscopia de lecho ungueal evidenció un patrón sugestivo de asociación a DM (fig. 3).
El electromiograma mostró un patrón miopático, con predominio proximal de miembros superiores. No se consideró necesario realizar biopsia muscular. Dada la elevada sensibilidad de la prueba para detectar neoplasia oculta, se realizó un PET-TC que fue normal.
Con el diagnóstico de DM, se instauró tratamiento con prednisona (1mg/kg/día). Tras 3 semanas, se observó mejoría notable de las lesiones cutáneas, pero persistía la debilidad muscular, añadiéndose disfagia. Analíticamente se incrementaron los niveles de CK hasta 3.800μ/l. Se añadió al tratamiento metotrexato semanal (hasta 20mg/semana SC) y un curso de inmunoglobulinas intravenosas a dosis altas. El paciente presentó mejoría de la fuerza muscular y de la deglución, así como desaparición de las pápulas de Gottron, con descenso paulatino de la CK.
DiscusiónLa asociación de MG a MII es muy infrecuente, habiéndose comunicado hasta la actualidad 26 casos3–9. Es importante conocer esta asociación ya que tiene implicaciones terapéuticas. A diferencia de los pacientes con MII, la MG cursa con debilidad fluctuante que empeora con la actividad y con el transcurso del día. Además, no es característica una auténtica fatigabilidad muscular en las MII, que es el dato clínico típico de la MG. En la mayoría de pacientes con MG, se afecta inicialmente la musculatura ocular, causando diplopia intermitente y ptosis palpebral, síntomas no observados en las MII. La debilidad muscular proximal es un síntoma común en la MG y en la DM, pudiendo aparecer en ambas síntomas bulbares, como disfagia o disartria.
El tratamiento de la MG incluye la utilización de anticolinesterásicos aunque no todos los pacientes responden. Como en las MII, los glucocorticoides, inmunosupresores e inmunomoduladores —inmunoglobulinas intravenosas— se usan frecuentemente en la MG. La pauta habitual de inicio de corticoides utilizada en la DM, es a dosis altas (1mg prednisona/kg/día) con posterior descenso gradual. Sin embargo, en la MG se recomienda iniciar con dosis bajas e incrementar progresivamente debido al riesgo de exacerbación de la debilidad muscular10.
En resumen, la MG puede preceder o complicar el curso de una MII. Conocer esta asociación es importante ya que requiere un manejo y tratamiento diferente, con una intensificación gradual de la terapia con glucocorticoides.
Responsabilidades éticasProtección de personas y animalesLos autores declaran que para esta investigación no se han realizado experimentos en seres humanos ni en animales.
Confidencialidad de los datosLos autores declaran que han seguido los protocolos de su centro de trabajo sobre la publicación de datos de pacientes.
Derecho a la privacidad y consentimiento informadoLos autores han obtenido el consentimiento informado de los pacientes y/o sujetos referidos en el artículo. Este documento obra en poder del autor de correspondencia.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.


